
1 Ximelagatran
2 Dabigatran
3 Argatroban
4
5
6
will be updated
- Argatroban
- Dabigatran
- Melagatran‡
- Ximelagatran‡
1
Ximelagatran

Ximelagatran
192939-46-1, EXANTAN-[(1R)-1-cyclohexyl-2-[(2S)-2-[[[[4- [(hydroxyamino)iminomethyl]phenyl]methyl]amino]carbonyl]-1- azetidinyl]-2-oxoethyl]-glycine, ethyl ester
| C24H35N5O5 | |
| MW | 473.6 |
CAS 260790-59-8 (MonoHBr)
CAS 260790-60-1 (Monomethanesulfonate)
ASTRAZENECA INNOVATOR
Ximelagatran (Exanta or Exarta, H 376/95) is an anticoagulant that has been investigated extensively as a replacement forwarfarin[1] that would overcome the problematic dietary, drug interaction, and monitoring issues associated with warfarin therapy. In 2006, its manufacturer AstraZeneca announced that it would withdraw pending applications for marketing approval after reports ofhepatotoxicity (liver damage) during trials, and discontinue its distribution in countries where the drug had been approved (Germany, Portugal, Sweden, Finland, Norway, Iceland, Austria, Denmark, France, Switzerland, Argentina and Brazil).[2]
Ximelagatran is an ester prodrug of melagatran, a potent, direct, and reversible thrombin inhibitor (Ki = 1.2 nM). While melagatran has poor oral bioavailability, ximelagatran displays good bioavailability resulting, in part, from rapid absorption at the gastrointestinal tract, as well as rapid onset of action.Ximelagatran is converted to melagatran by reduction and hydrolysis at the liver and other tissues. It is used as an anticoagulant in a variety of situations, including thromboembolic disorders, stroke prevention in atrial fibrillation, and therapy in vein thrombosis
Method of action
Ximelagatran, a direct thrombin inhibitor,[3] was the first member of this class that can be taken orally. It acts solely by inhibiting the actions of thrombin. It is taken orally twice daily, and rapidly absorbed by the small intestine. Ximelagatran is a prodrug, being converted in vivo to the active agent melagatran. This conversion takes place in the liver and many other tissues throughdealkylation and dehydroxylation (replacing the ethyl and hydroxyl groups with hydrogen).Uses
Ximelagatran was expected to replace warfarin and sometimes aspirin and heparin in many therapeutic settings, including deep venous thrombosis, prevention of secondary venous thromboembolism and complications of atrial fibrillation such as stroke. The efficacy of ximelagatran for these indications had been well documented,[4][5][6] except for non valvular atrial fibrillation.An advantage, according to early reports by its manufacturer, was that it could be taken orally without any monitoring of its anticoagulant properties. This would have set it apart from warfarin and heparin, which require monitoring of the international normalized ratio (INR) and the partial thromboplastin time (PTT), respectively. A disadvantage recognised early was the absence of an antidote in case acute bleeding develops, while warfarin can be antagonised by vitamin K and heparin by protamine sulfate.
Side-effects
Ximelagatran was generally well tolerated in the trial populations, but a small proportion (5-6%) developed elevated liver enzymelevels, which prompted the FDA to reject an initial application for approval in 2004. The further development was discontinued in 2006 after it turned out hepatic damage could develop in the period subsequent to withdrawal of the drug. According to AstraZeneca, a chemically different but pharmacologically similar substance, AZD0837, is undergoing testing for similar indications.[2]Melagatran synthesis

Sobrera, L. A.; Castaner, J.; Drugs Future, 2002, 27, 201.
SYNTHESIS

SYNTHESIS

SYNTHESIS



……
WO 1997023499/http://www.google.com/patents/EP0869966A1?cl=en…………
References
- Hirsh J, O’Donnell M, Eikelboom JW (July 2007). “Beyond unfractionated heparin and warfarin: current and future advances”. Circulation 116 (5): 552–560.doi:10.1161/CIRCULATIONAHA.106.685974. PMID 17664384.
- “AstraZeneca Decides to Withdraw Exanta” (Press release). AstraZeneca. February 14, 2006. Retrieved 2012-07-16.
- Ho SJ, Brighton TA (2006). “Ximelagatran: direct thrombin inhibitor”. Vasc Health Risk Manag 2 (1): 49–58. doi:10.2147/vhrm.2006.2.1.49. PMC 1993972.PMID 17319469.
- Eriksson, H; Wahlander K; Gustafsson D; Welin LT; Frison L; Schulman S; THRIVE Investigators (January 2003). “A randomized, controlled, dose-guiding study of the oral direct thrombin inhibitor ximelagatran compared with standard therapy for the treatment of acute deep vein thrombosis: THRIVE I”. Journal of Thrombosis and Haemostasis 1 (1): 41–47. doi:10.1046/j.1538-7836.2003.00034.x. PMID 12871538.
- Francis, CW; Berkowitz SD, Comp PC, Lieberman JR, Ginsberg JS, Paiement G, Peters GR, Roth AW, McElhattan J, Colwell CW Jr; EXULT A Study Group (October 2003). “Comparison of ximelagatran with warfarin for the prevention of venous thromboembolism after total knee replacement”. New England Journal of Medicine 349 (18): 1703–1712.doi:10.1056/NEJMoa035162. PMID 14585938.
- Schulman, S; Wåhlander K; Lundström T; Clason SB; Eriksson H; THRIVE III investigators (October 2003). “Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran”. New England Journal of Medicine 349 (18): 1713–1721. doi:10.1056/NEJMoa030104. PMID 14585939.
 |
|
| Systematic (IUPAC) name | |
|---|---|
ethyl 2-[[(1R)-1-cyclohexyl-2-
[(2S)-2-[[4-(N’-hydroxycarbamimidoyl) phenyl]methylcarbamoyl]azetidin-1-yl]- 2-oxo-ethyl]amino]acetate |
|
| Clinical data | |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 20% |
| Metabolism | None |
| Biological half-life | 3-5h |
| Excretion | Renal (80%) |
| Identifiers | |
| CAS Registry Number | 192939-46-1 |
| ATC code | B01AE05 |
| PubChem | CID: 9574101 |
| IUPHAR/BPS | 6381 |
| DrugBank | DB04898 |
| ChemSpider | 7848559 |
| UNII | 49HFB70472 |
| KEGG | D01981 |
| ChEMBL | CHEMBL522038 |
| Chemical data | |
| Formula | C24H35N5O5 |
| Molecular mass | 473.57 g·mol−1 (429 g/mol after conversion) |

2
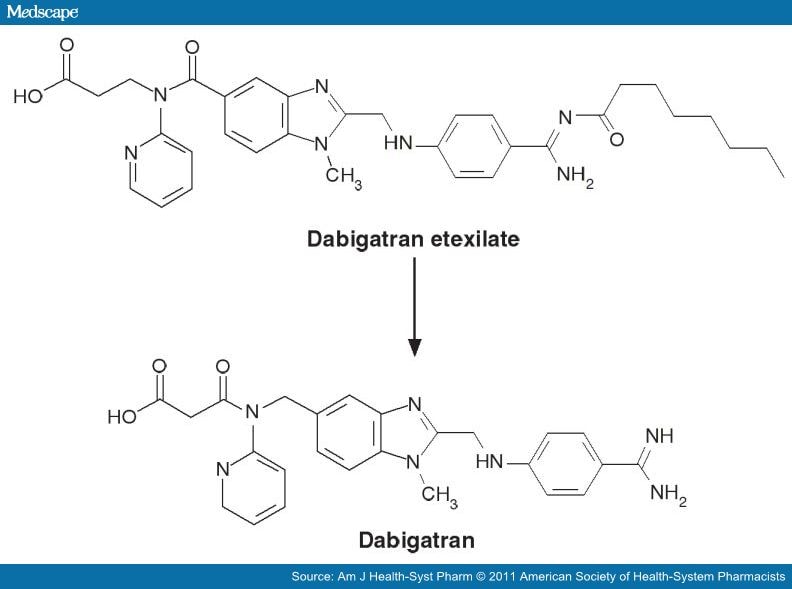
DABIGATRAN

Dabigatran (Pradaxa in Australia, Canada, Europe and USA, Prazaxa in Japan) is an oral anticoagulant from the class of the direct thrombin inhibitors. It is being studied for various clinical indications and in some cases it offers an alternative towarfarin as the preferred orally administered anticoagulant ("blood thinner") since it cannot be monitored by blood tests forinternational normalized ratio (INR) monitoring while offering similar results in terms of efficacy. There is no specific way to reverse the anticoagulant effect of dabigatran in the event of a major bleeding event,[2][3] unlike warfarin,[4] although a potential dabigatran antidote (pINN: idarucizumab) is undergoing clinical studies.[5] It was developed by the pharmaceutical company Boehringer Ingelheim.
Medical uses
Dabigatran is used to prevent strokes in those with atrial fibrillation (afib) due to non heart valve causes, as well as deep venous thrombosis (DVT) and pulmonary embolism (PE) in persons who have been treated for 5–10 days with parenteral anticoagulant (usually low molecular weight heparin), and to prevent DVT and PE in some circumstances.[6]
It appears to be as effective as warfarin in preventing nonhemorrhagic strokes and embolic events in those with afib not due to valve problems.[7]

Contraindications
Dabigatran is contraindicated in patients who have active pathological bleeding since dabigatran can increase bleeding risk and can also cause serious and potentially life-threatening bleeds.[8] Dabigatran is also contraindicated in patients who have a history of serious hypersensitivity reaction to dabigatran (e.g. anaphylaxis or anaphylactic shock).[8] The use of dabigatran should also be avoided in patients with mechanical prosthetic heart valve due to the increased risk of thromboembolic events (e.g. valve thrombosis, stroke, and myocardial infarction) and major bleeding associated with dabigatran in this population.[8][9][10]
Adverse effects
The most commonly reported side effect of dabigatran is GI upset. When compared to people anticoagulated with warfarin, patients taking dabigatran had fewer life-threatening bleeds, fewer minor and major bleeds, including intracranial bleeds, but the rate of GI bleeding was significantly higher. Dabigatran capsules contain tartaric acid, which lowers the gastric pH and is required for adequate absorption. The lower pH has previously been associated with dyspepsia; some hypothesize that this plays a role in the increased risk of gastrointestinal bleeding.[11]
A small but significantly increased risk of myocardial infarctions (heart attacks) has been noted when combining the safety outcome data from multiple trials.[12]
Reduced doses should be used in those with poor kidney function.[13]
Pharmacokinetics
Dabigatran has a half-life of approximately 12-14 h and exert a maximum anticoagulation effect within 2-3 h after ingestion.[14] Fatty foods delay the absorption of dabigatran, although the bio-availability of the drug is unaffected.[1] One study showed that absorption may be moderately decreased if taken with a proton pump inhibitor.[15] Drug excretion through P-glycoprotein pumps is slowed in patients taking strong p-glycoprotein pump inhibitors such as quinidine, verapamil, and amiodarone, thus raising plasma levels of dabigatran.[16]
History
Dabigatran (then compound BIBR 953) was discovered from a panel of chemicals with similar structure to benzamidine-based thrombin inhibitor α-NAPAP (N-alpha-(2-naphthylsulfonylglycyl)-4-amidinophenylalanine piperidide), which had been known since the 1980s as a powerful inhibitor of various serine proteases, specifically thrombin, but also trypsin. Addition of ethyl ester and hexyloxycarbonyl carbamide hydrophobic side chains led to the orally absorbed prodrug, BIBR 1048 (dabigatran etexilate).[17]
On March 18, 2008, the European Medicines Agency granted marketing authorisation for Pradaxa for the prevention of thromboembolic disease following hip or knee replacement surgery and for non-valvular atrial fibrillation.[18]
The National Health Service in Britain authorised the use of dabigatran for use in preventing blood clots in hip and knee surgery patients. According to a BBC article in 2008, Dabigatran was expected to cost the NHS £4.20 per day, which was similar to several other anticoagulants.[19]
Pradax received a Notice of Compliance (NOC) from Health Canada on June 10, 2008,[20] for the prevention of blood clots in patients who have undergone total hip or total knee replacement surgery. Approval for atrial fibrillation patients at risk of stroke came in October 2010.[21][22]
The U.S. Food and Drug Administration (FDA) approved Pradaxa on October 19, 2010, for prevention of stroke in patients with non-valvular atrial fibrillation.[23][24][25][26] The approval came after an advisory committee recommended the drug for approval on September 20, 2010[27] although caution is still urged by some outside experts.[28]
On February 14, 2011, the American College of Cardiology Foundation and American Heart Association added dabigatran to their guidelines for management of non-valvular atrial fibrillation with a class I recommendation.[29]
In May 2014 the FDA reported the results of a large study comparing dabigatran to warfarin in 134,000 Medicare patients. The Agency concluded that dabigatran is associated with a lower risk of overall mortality, ischemic stroke, and bleeding in the brain than warfarin. Gastrointestinal bleeding was more common in those treated with dabigatran than in those treated with warfarin. The risk of heart attack was similar between the two drugs. The Agency reiterated its opinion that dabigatran's overall risk/benefit ratio is favorable.[30]
On July 26, 2014, the British Medical Journal (BMJ) published a series of investigations that accused Boehringer of withholding critical information about the need for monitoring to protect patients from severe bleeding, particularly in the elderly. Review of internal communications between Boehringer researchers and employees, the FDA and the EMA revealed that Boehringer researchers found evidence that serum levels of dabigatran vary widely. The BMJ investigation suggested that Boehringer had a financial motive to withhold this concern from regulatory health agencies because the data conflicted with their extensive marketing of dabigatran as an anticoagulant that does not require monitoring.[31][32]
Research
In August 2015, an article found that idarucizumab was able to reverse the anticoagulation effects of dabigatran within minutes.[33]
References
- Pradaxa Full Prescribing Information. Boehringer Ingelheim. October 2010.
- Eerenberg, ES; Kamphuisen, PW; Sijpkens, MK; Meijers, JC; Buller, HR; Levi, M (2011-10-04). "Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects". Circulation 124(14): 1573–9. doi:10.1161/CIRCULATIONAHA.111.029017. PMID 21900088. Retrieved 2012-03-15.
- van Ryn J, Stangier J, Haertter S, Liesenfeld KH, Wienen W, Feuring M, Clemens A (Department of Drug Discovery Support, Boehringer Ingelheim Pharma) (Jun 2010)."Dabigatran etexilate--a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity". Thrombosis and Haemostasis103 (6): 1116–27. doi:10.1160/TH09-11-0758. PMID 20352166. Retrieved2012-03-15.
Although there is no specific antidote to antagonise the anticoagulant effect of dabigatran, due to its short duration of effect drug discontinuation is usually sufficient to reverse any excessive anticoagulant activity.
- Hanley JP, J P (Nov 2004). "Warfarin reversal". Journal of Clinical Pathology 57 (11): 1132–9. doi:10.1136/jcp.2003.008904. PMC 1770479. PMID 15509671.
- "Boehringer Ingelheim’s Investigational Antidote for Pradaxa® (dabigatran etexilate mesylate) Receives FDA Breakthrough Therapy Designation" (Press release). Ridgefield, CT: Boehringer Ingelheim’. 2014-06-26. Retrieved 2014-07-26.
- http://www.drugs.com/pro/pradaxa.html Pradaxa
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). "Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.". Thrombosis 2013: 640723. doi:10.1155/2013/640723. PMC 3885278.PMID 24455237.
- Pradaxa (dabigatran etexilate mesylate) Prescribing Information:http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=ba74e3cd-b06f-4145-b284-5fd6b84ff3c9#Section_5.4, accessed October 29, 2014.
- "FDA Drug Safety Communication: Pradaxa (dabigatran etexilate mesylate) should not be used in patients with mechanical prosthetic heart valves". U.S. Food and Drug Administration (FDA). Retrieved October 29, 2014.
- Eikelboom, JW; Connolly, SJ; Brueckmann, M et al. (September 2013). "Dabigatran versus Warfarin in Patients with Mechanical Heart Valves". N Engl J Med 369: 1206–1214.doi:10.1056/NEJMoa1300615. PMID 23991661.
- ML Blommel et al. (2011). "Dabigatran etexilate: A novel oral direct thrombin inhibitor".Am J Health Syst Pharm 68 (16): 1506–19. doi:10.2146/ajhp100348. PMID 21817082.
- Uchino K, Hernandez AV; Hernandez (2012). "Dabigatran associated with higher risk of acute coronary events - meta-analysis of noninferiority randomized controlled trials".Arch. Intern. Med. Online first (5): 397–402. doi:10.1001/archinternmed.2011.1666.PMID 22231617.
- 18/12/2014 Pradaxa -EMEA/H/C/000829 -II/0073
- Chongnarungsin D; Ratanapo S; Srivali N; Ungprasert P; Suksaranjit P; Ahmed S; Cheungpasitporn W (2012). "In-Depth Review of Stroke Prevention in Patients with Non-Valvular Atrial Fibrillation". Am. Med. J. 3 (2): 100. doi:10.3844/amjsp.2012.100.103.
- Stangier J, Eriksson BI, Dahl OE et al. (May 2005). "Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement". J Clin Pharmacol 45 (5): 555–63.doi:10.1177/0091270005274550. PMID 15831779.
- "Pradaxa Summary of Product Characteristics". European Medicines Agency.
- Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W; Nar; Priepke; Ries; Stassen; Wienen (April 2002). "Structure-based design of novel potent nonpeptide thrombin inhibitors". J Med Chem 45 (9): 1757–66. doi:10.1021/jm0109513.PMID 11960487. Lay summary.
- "Pradaxa EPAR". European Medicines Agency. Retrieved 2011-01-30.
- "Clot drug 'could save thousands'". BBC News Online. 2008-04-20. Retrieved2008-04-21.
- "Summary Basis of Decision (SBD): Pradax" Health Canada. 2008-11-06.
- Kirkey, Sharon (29 October 2010). "Approval of new drug heralds 'momentous' advance in stroke prevention". Montreal Gazette. Retrieved 29 October 2010.
- "Pradax (Dabigatran Etexilate) Gains Approval In Canada For Stroke Prevention In Atrial Fibrillation" Medical News Today. 28 October 2010.
- Connolly, SJ; Ezekowitz, MD; Yusuf, S et al. (September 2009). "Dabigatran versus warfarin in patients with atrial fibrillation" (PDF). N Engl J Med 361 (12): 1139–51.doi:10.1056/NEJMoa0905561. PMID 19717844.
- Turpie AG (January 2008). "New oral anticoagulants in atrial fibrillation". Eur Heart J 29(2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
- "Boehringer wins first US OK in blood-thinner race". Thomson Reuters. 2010-10-19. Retrieved 2010-10-20.
- "FDA approves Pradaxa to prevent stroke in people with atrial fibrillation". U.S. Food and Drug Administration (FDA). 2010-10-19.
- Shirley S. Wang (2010-09-20). "New Blood-Thinner Recommended by FDA Panel". The Wall Street Journal. Retrieved 2010-10-20.
- Merli G, Spyropoulos AC, Caprini JA; Spyropoulos; Caprini (August 2009). "Use of emerging oral anticoagulants in clinical practice: translating results from clinical trials to orthopedic and general surgical patient populations". Ann Surg 250 (2): 219–28.doi:10.1097/SLA.0b013e3181ae6dbe. PMID 19638915.
- Wann LS, Curtis AB, Ellenbogen KA, Estes NA, Ezekowitz MD, Jackman WM, January CT, Lowe JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK; Curtis; Ellenbogen; Estes Na; Ezekowitz; Jackman; January; Lowe; Page; Slotwiner; Stevenson; Tracy; Fuster; Rydén; Cannom; Crijns; Curtis; Ellenbogen; Halperin; Kay; Le Heuzey; Lowe; Olsson; Prystowsky; Tamargo; Wann; Jacobs; Anderson; Albert et al. (March 2011). "2011 ACCF/AHA/HRS Focused Update on the Management of Patients With Atrial Fibrillation (Update on Dabigatran): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines". Circulation123 (10): 1144–50. doi:10.1161/CIR.0b013e31820f14c0. PMID 21321155.
- "FDA Drug Safety Communication: FDA study of Medicare patients finds risks lower for stroke and death but higher for gastrointestinal bleeding with Pradaxa (dabigatran) compared to warfarin".
- Cohen, D (July 2014). "Dabigatran: how the drug company withheld important analyses".BMJ 349: g4670. doi:10.1136/bmj.g4670. PMID 25055829.
- Moore TJ, Cohen MR, Mattison DR; Cohen; Mattison (July 2014). "Dabigatran, bleeding, and the regulators". BMJ 349: g4517. doi:10.1136/bmj.g4517. PMID 25056265.
- Pollack, Charles V.; Reilly, Paul A.; Eikelboom, John; Glund, Stephan; Verhamme, Peter; Bernstein, Richard A.; Dubiel, Robert; Huisman, Menno V.; Hylek, Elaine M. (2015-01-01)."Idarucizumab for Dabigatran Reversal". New England Journal of Medicine 373 (6).doi:10.1056/nejmoa1502000.
 | |
| Systematic (IUPAC) name | |
|---|---|
| Ethyl N-[(2-{[(4-{N '-[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl]-N-2-pyridinyl-β-alaninate | |
| Clinical data | |
| Trade names | Pradaxa, Pradax, Prazaxa |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | oral |
| Pharmacokinetic data | |
| Bioavailability | 3–7%[1] |
| Protein binding | 35%[1] |
| Biological half-life | 12–17 hours[1] |
| Identifiers | |
| CAS Registry Number | 211915-06-9 |
| ATC code | B01AE07 |
| PubChem | CID: 6445226 |
| DrugBank | DB06695 |
| ChemSpider | 4948999 |
| ChEMBL | CHEMBL539697 |
| Chemical data | |
| Formula | C34H41N7O5 |
| Molecular mass | 627.734 g/mol |
External links
- Pradaxa.com. Boehringer Ingelheim.
- dabigatran.com. Boehringer Ingelheim.
- Pradaxa For U.S. Health Care Professionals. Boehringer Ingelheim.
- Pradaxa Prescribing Information. Boehringer Ingelheim.
- Pradaxa Medication Guide. Boehringer Ingelheim.
- Dabigatran. MedlinePlus. United States National Library of Medicine (NLM).
- Dabigatran. Drug Information Portal. United States National Library of Medicine (NLM).
The chemical name for dabigatran etexilate mesylate, a direct thrombininhibitor, is β-Alanine, N-[[2-[[[4-[[[(hexyloxy)carbonyl]amino]iminomethyl] phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl-,ethyl ester, methanesulfonate. The empirical formula is C34H41N7O5 • CH4O3S and the molecular weight is 723.86 (mesylate salt), 627.75 (free base). The structural formula is:
 |
Dabigatran etexilate mesylate is a yellow-white to yellow powder. A saturated solution in pure water has a solubility of 1.8 mg/mL. It is freely soluble in methanol, slightly soluble in ethanol, and sparingly soluble in isopropanol.
The 150 mg capsule for oral administration contains 172.95 mg dabigatran etexilate mesylate, which is equivalent to 150 mg of dabigatran etexilate, and the following inactive ingredients: acacia, dimethicone, hypromellose, hydroxypropyl cellulose, talc, and tartaric acid. The capsule shell is composed of carrageenan, FD&C Blue No. 2 (150 mg only), FD&C Yellow No. 6, hypromellose, potassium chloride, titanium dioxide, and black edible ink. The 75 mg capsule contains 86.48 mg dabigatran etexilate mesylate, equivalent to 75 mg dabigatran etexilate, and is otherwise similar to the 150 mg capsule.
/////////Dabigatran, Pradaxa

- Dabigatran etexilate (a compound of formula (I)) is the international commonly accepted non-proprietary name for ethyl 3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate, which has an empirical formula of C34H41N7O5 and a molecular weight of 627.73.

- Dabigatran etexilate is the pro-drug of the active substance, dabigatran, which has a molecular formula C25H25N7O3 and molecular mass 471.51. The mesylate salt (1:1) of dabigatran etexilate is known to be therapeutically useful and is commercially marketed as oral hard capsules in the United States and in Europe under the trade mark Pradaxa™ for the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation. Additionally, it is also marketed in Europe under the same trade mark for the primary prevention of venous thromboembolic events in adult patients who have undergone elective total hip replacement surgery or total knee replacement surgery.
- Dabigatran etexilate was first described in U.S. Patent No. 6,087,380 , according to which the synthesis of dabigatran etexilate was carried out in three synthetic steps (see Scheme 1). Example 58 describes the condensation between ethyl 3-{[3-amino-4-(methylamino)benzoyl](pyridin-2-yl)amino}propanoate (compound II) and N-(4-cyanophenyl)glycine (compound III) in the presence of N,N'-carbonyldiimidazole (CDI) in tetrahydrofuran to give the hydrochloride salt of ethyl 3-{[(2-{[(4-cyanophenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (compound IV), which is subsequently reacted with ethanolic hydrochloric acid, ethanol and ammonium carbonate to give the hydrochloride salt of ethyl 3-{[(2-{[(4-carbamimidoylphenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (compound V). Finally, example 113 describes the reaction between compound V and n-hexyl chloroformate (compound VI), in the presence of potassium carbonate, in a mixture of tetrahydrofuran and water, to give dabigatran etexilate after work-up and chromatographic purification. However, no information is given about the purity of the isolated dabigatran etexilate.


- U.S. Patent No. 7,202,368 describes an alternative process for the synthesis of dabigatran etexilate (see Scheme 2). Example 3 describes the condensation between ethyl 3-{[3-amino-4-(methylamino)benzoyl](pyridin-2-yl)amino}propanoate (compound II) and 2-[4-(1,2,4-oxadiazol-5-on-3-yl)phenylamino]acetic acid (compound VII) in the presence of a coupling agent such as N,N'-carbonyldiimidazole (CDI), propanephosphonic anhydride (PPA), or pivaloyl chloride, to give ethyl 3-{[(2-{[(4-{1,2,4-oxadiazol-5-on-3-yl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (compound VIII), which is subsequently hydrogenated (Example 4) in the presence of a palladium catalyst to give ethyl 3-{[(2-{[(4-carbamimidoylphenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (compound V). Then, Example 5 describes the acylation of compound V with n-hexyl chloroformate (compound VI) to give dabigatran etexilate. Finally, Example 6 describes the conversion of dabigatran etexilate into its mesylate salt. Although the patent describes the HPLC purities of intermediate compounds II, VII, VIII and V, no information is given neither about the purity of the isolated dabigatran etexilate nor about its mesylate salt.


- European Patent Applications EP 1966171A and EP 1968949Adescribe similar processes for the synthesis of dabigatran etexilate to that depicted in Scheme 2, but without isolating some of the intermediate compounds. HPLC purities higher than 99% are described for both dabigatran etexilate (see Examples 6B and 6C ofEP 1966171A ) and its mesylate salt (see Example 9 ofEP 1966171A and Example 7 ofEP 1968949A). However, no information is given about the structure of the impurities present in dabigatran etexilate and / or its mesylate salt.
- PCT Patent Application WO 2010/045900 describes the synthesis of dabigatran etexilate mesylate with 99.5% purity by HPLC (Examples 3 and 4) by following a similar synthetic process to that described in Scheme 1. However, no information is given about the structure of the impurities present in the mesylate salt of dabigatran etexilate.
- The Committee for Medicinal Products for Human use (CHMP) assessment report for Pradaxa (i.e. dabigatran etexilate mesylate salt 1:1) reference EMEA/174363/2008, as published in the European Medicines Agency website on 23/04/2008, describes (page 8) that the proposed specifications for impurities in the active substance are for some specified impurities above the qualification threshold of the ICH guideline "Impurities in new drug substances", i.e. above 0.15%. However, no information is given about the structure of the impurities present in the mesylate salt of dabigatran etexilate.
..............
Patent
There is still further provided by the present invention a process of preparing dabigatran etexilate mesylate, which process comprises the following synthetic steps:

wherein X is a leaving group, such as chloro.
Typically, intermediate (I) is prepared, preferably as a hydrochloride salt, by the following intermediate steps.

Example 1: Synthesis of dabigatran etexilate mesylate
- The overall synthetic scheme, and associated reagents, is as follows.


- 300 g (1.49 mol) of 4-chloro-3-nitrobenzoic acid were suspended in 769 g of a 25-30% aqueous solution of methylamine. After heating to reflux temperature, a clear solution was obtained. The solution was kept at reflux temperature for 2 hours and total consumption of 4-chloro-3-nitrobenzoic acid was checked by TLC. The solution was cooled to room temperature, and pH was adjusted to about 1 by addition of 2M aqueous sulphuric acid. Precipitation of a yellow solid was observed, which was isolated by filtration. The filtered cake was washed with water and subsequently with methanol to obtain 331 g of wet 4-(methylamino)-3-nitrobenzoic acid as a yellow powder. Purity (HPLC, method 2): 99.1 %.

- 75.2 g (0.80 mol) of 2-aminopyridine and 88.0 g (0.88 mol) of ethyl acrylate were dissolved in 20 mL of acetic acid. The mixture was heated to 80°C and stirred for 24 hours at the same temperature. Solvent was removed under vacuum, and the title compound was isolated by vacuum distillation (b.p. 160-172°C, 10-15 mmHg) to obtain 77.0 g of ethyl 3-(2-pyridylamino)propionate as a white solid. Yield: 49.6 %.

- 50 g (0.25 mol) of 4-(methylamino)-3-nitrobenzoic acid as obtained in step (a) were suspended in a mixture of 459.2 g of thionyl chloride and 3 mL of N,N-dimethylformamide. The mixture was stirred at reflux temperature for 45 minutes. Excess thionyl chloride was removed by vacuum distillation. The residue was dissolved in 300 mL of toluene, which was subsequently removed by vacuum distillation to remove completely any residual thionyl chloride. The brownish crystalline residue obtained was dissolved in 280 mL of tetrahydrofuran at 60°C. At this point, 35.1 g of triethylamine were added to the solution. Then, a solution of 45 g (0.23 mol) of ethyl 3-(2-pyridylamino)propanoate as obtained in step (b) in 95 mL of tetrahydrofuran was added dropwise over the reaction mixture, keeping the temperature at about 30°C. The resulting mixture was stirred overnight at room temperature. Solvent was removed by vacuum distillation, and the residue was dissolved in 1 L of dichloromethane. The resulting solution was washed with 500 mL of water, 500 mL of 2M hydrochloric acid, 500 mL of saturated sodium bicarbonate and 500 mL of water. The organic phase was dried with anhydrous sodium sulfate and concentrated under vacuum. The residue was dissolved with 600 mL of ethyl acetate, and dry hydrogen chloride was bubbled into the solution until precipitation was completed. The solid was isolated by filtration and dried to obtain 63 g of the title compound, which was recrystallized in a mixture of 450 mL of ethanol and 50 mL of acetonitrile at reflux temperature. After cooling to 10°C, solid was isolated by filtration and dried to yield 44.7 g of ethyl 3-{[{1-(methylamino)-2-nitrophen-4-yl}carbonyl](pyridyn-2-yl)amino}propanoate hydrochloride as a yellow solid. Yield: 47.2 %. Purity (HPLC, method 1): 97.6 %.

- 82.2 g (0.20 mol) of ethyl 3-{[{1-(methylamino)-2-nitrophen-4-yl}carbonyl](pyridyn-2-yl)amino}propanoate hydrochloride as obtained in step (c) were suspended in 1.1 L of isopropanol, in the presence of 126.7 g of ammonium formate and 17.5 g of a 5 % Pd/C catalyst (55% water content). The reaction mixture was stirred at reflux temperature for 2.5 hours. After cooling to room temperature, the catalyst was removed by filtration, the filtrate was concentrated under vacuum, and the residue was dissolved in 1.5 L of ethyl acetate. The resulting solution was washed with 800 mL of saturated sodium bicarbonate and with 800 mL of water. The organic phase was dried with anhydrous sodium sulfate and was concentrated under vacuum to yield 44 g of ethyl 3-{[{2-amino-1-(methylamino)phen-4-yl}carbonyl](pyridyn-2-yl)amino}propanoate as a dark oil. Yield: 63.9 %. Purity (HPLC, method 2): 90.8 %.

- 54.0 g (0.46 mol) of 4-aminobenzonitrile and 106.5 g (0.92 mol) of sodium chloroacetate were suspended in 750 mL of water, and the resulting mixture was stirred at reflux temperature for 4 hours. After cooling to room temperature, pH was adjusted to 8-9 with sodium bicarbonate. The resulting solution was washed with 2 x 200 mL of ethyl acetate, and 5M hydrochloric acid was added to the aqueous phase until pH=3. The precipitated solid was isolated by filtration, washed with 100 mL of water and dried to yield 57.1 g of 2-(4-cyanophenylamino)acetic acid as an off-white solid. Yield: 70.9 %. Purity (HPLC, method 3): 88.4 %.
- [0081]

- 25.7 g (0.15 mol) of 2-(4-cyanophenylamino)acetic acid as obtained in step (e) and 22.8 g (0.14 mol) of 1,1'-carbonyldiimidazole were suspended in 720 mL of tetrahydrofuran. The mixture was stirred at reflux temperature for 1 hour. Then, a solution of 44.0 g (0.13 mol) of ethyl 3-{[{2-amino-1-(methylamino)phen-4-yl}carbonyl](pyridyn-2-yl)amino}propanoate as obtained in step (d) in 180 mL of tetrahydrofuran was added dropwise over the reaction mixture. The resulting mixture was stirred overnight at reflux temperature, and the solvent was removed by distillation under vacuum. The resulting residue was dissolved in 486 mL of acetic acid and heated to reflux temperature for 1 hour. After cooling to room temperature, solvent was removed by distillation under vacuum. The resulting residue was dissolved in 450 mL of ethyl acetate, and the solution was washed with 450 mL of water. The organic phase was dried with anhydrous sodium sulfate and heated to 50-60°C. At this temperature, 15.1 g (0.17 mol) of oxalic acid were added, and the resulting mixture was stirred for 1 hour at 50-60°C. After cooling to room temperature, the precipitated solid was filtered and dried under vacuum, to yield 47.7 g of ethyl 3-{[(2-{[(4-cyanophenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate oxalate as a brownish solid. Yield: 64.8 %. Purity (HPLC, method 1): 87.9 %

- [0084]47.7 g (83 mmol) of ethyl 3-{[(2-{[(4-cyanophenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate oxalate as obtained in step (f) and 21.8 g of p-toluenesulfonic acid were suspended in 142 g of a 10M hydrogen chloride solution in ethanol. The mixture was stirred at room temperature for 24 hours. At this point, 400 mL of ethanol were added and the resulting mixture was cooled to 0°C. Ammonia gas was bubbled at this temperature until formation of precipitate was completed. The mixture was stirred at 10°C for 2 hours, and then was stirred at room temperature overnight. Solvent was removed by distillation under vacuum. The residue was dissolved in a mixture of 400 mL of ethanol, 400 mL of water and 2.3 g of sodium hydroxide at 55°C, and was stirred at this temperature for 45 minutes. After cooling to 10°C, the mixture was stirred at this temperature for 1 hour. The solid was removed by filtration and discarded. The mother liquors were concentrated under vacuum to remove ethanol. The precipitated solid was isolated by filtration, washed with 200 mL of water and with 2 x 100 mL of acetone, to yield 34.7 g of ethyl 3-{[(2-{[(4-{carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate as an off-white solid. Yield: 83.4 %. Purity (HPLC, method 3): 83 %.

- 33.7 g (67 mmol) of ethyl 3-{[(2-{[(4-{carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate as obtained in step (g) and 24.7 g of potassium carbonate were suspended in a mixture of 280 mL of water and 1.4 L of tetrahydrofuran. After stirring at room temperature for 15 minutes, 9.2 g (56 mmol) of hexyl chloroformate were added dropwise. The resulting mixture was stirred at room temperature for 1 hour. The organic phase was extracted, washed with 400 mL of brine and dried with anhydrous sodium sulfate. The solvent was removed under vacuum, and the resulting solid was purified by column chromatography eluting with ethyl acetate, to yield 24.9 g of dabigatran etexilate as an off-white solid. Yield: 71.0 %. Purity (HPLC, method 1): 96.3 %.
- 18.7 g (30 mmol) of dabigatran etexilate as obtained in step (h) were suspended in 103 g of acetone. The mixture was heated to 45°C. After cooling to 36°C, a solution of 2.83 g of methanesulfonic acid in 11.6 g of acetone at 0°C was added dropwise over the reaction mixture. The reaction was stirred at 23-33°C for 90 minutes and at 17-23°C for 60 minutes. The resulting solid was isolated by filtration, washed with 97 mL of acetone and dried at 50°C under vacuum, to yield 18.7 g of dabigatran etexilate mesylate as a pale yellow solid. Yield: 86.7 %. Purity (HPLC, method 1): 98.8 %.
................
PATENT
One of the advanced intermediates during the production of dabigatran is the substance of formula VI.

VI
The compound of formula VI is prepared by a reaction of substance IV with reagent V as shown in Scheme 1.

Scheme 1
The procedure described in WO 9837075 produces compound VI in the form of its base or acetate. Both these products require chromatographic purification, which is very difficult to apply in the industrial scale. This purification method burdens the process economy very much and has a negative impact on the yield.
In the next stage acidic hydrolysis of the nitrile function of compound VI and a reaction with ammonium carbonate is performed to produce the substance of formula VII. The reaction is shown in Scheme 2.

Vl VII
Scheme 2 The procedure in accordance with WO 9837075 produces substance VII in the monohydro chloride form.
When reproducing the procedure of WO 9837075 we found out, in line with WO 9837075, that compound VII prepared by this method required subsequent chromatographic purification as it was an oily substance with a relatively high content of impurities. We did not manage to find a solvent that would enable purification of this substance by crystallization.
The last stage is a reaction of intermediate VII with hexyl chloroformate producing dabigatran and its transformation to a pharmaceutically acceptable salt; in the case of the above mentioned patent application it is the methanesulfonate.
Scheme 3.
EtOH



DABIGATRAN
Example 3: Preparation of dabigatran mesylate
To 9.1 g of compound VII-2HC1 (0.016 mol) 270 ml of chloroform and 9 ml (0.064 mol) of triethylamine are added. Then, a solution of 3.1 ml (0.018 mol) of hexyl chloroformate in chloroform is added dropwise at the laboratory temperature. After one hour the reaction mixture is shaken with brine and the organic layer is separated, which is dried with sodium sulfate and concentrated. The obtained evaporation residue is crystallized from ethyl acetate. Yield: 8.6 g (86%)
This product is dissolved in acetone and an equimolar amount of methanesulfonic acid is added dropwise. The separated precipitate is aspirated and dried at the laboratory temperature. Yield: 75%; content according to HPLC: 99.5%. 27
Example 4:
Preparation of dabigatran mesylate
9 g of compound VII-HCl (0.017 mol) were dissolved in 300 ml of chloroform. 6, ml of triethylamine were added to this solution and then a solution of 3.4 ml (0.02 mol) of hexyl chloroformate in chloroform was added dropwise. After one hour the reaction mixture is shaken with brine, the organic layer is separated, which is dried with sodium sulfate and concentrated. The obtained evaporation residue is crystallized from ethyl acetate. Yield: 9.6 g (90%)
This product is dissolved in acetone and an equimolar amount of methanesulfonic acid is added dropwise. The separated evaporation residue is aspirated and dried at the laboratory temperature. Yield: 73%; content according to HPLC: 99.5%.
....................
PATENT
DabigatranEtexilateMesylate chemically know as N-[[2-[[[4-[[[(hexyloxy) carbonyl] amino]-iminomethyl] phenyl] amino] . methyl]-l -methyl-lH- benzimidazol-5-yl] carbonyI]-N-2- pyridinyl-beta-Alanine ethyl ester methanesulfonate having the formula I as provided below,

Formula I
is a direct thrombin inhibitor having anti - coagulant activity when administered orally.
DabigatranEtexilate is first time reported in the US patent 6087380 (hereinafter referred as US'380) in which the process fo the preparation of DabigatranEtexilate is disclosed in the Example 49, 58a and Example 59, said process for the preparation of DabigatranEtexilate is depicted below:

Dabigatran etexilate
In accordance to the process in the Patent US'380 the substance requires complex purifying operations, such as chromatography for the production of high- quality API. Further the chromatographic purification is expensive and difficult to implement in large scale. The impurity in the Dabigatran single prodrug and Dabigatran Etexilate affects the purity of the final product DabigatranEtexilateMesylate.. Hence there is a necessity to maintain the purity level of every intermediate involved in the preparation of DabigatranEtexilateMesylate.
The patent application US201 1082299 discloses a process for the preparation Dabigatran from 3- ([2-[(4-cyanophenyl amino)-methyl]- l-methyl- l H-benzimidazole-5-carbonyl]-pyridin-2-yl-amino) ethyl propionate oxalate as one of the intermediate in order to overcome the problem of the process depicted in the product pate

The patent US81 19810 discloses the process for the preparation Dabigatran from 3- ([2-[(4-cyanophenylamino)-methyl]-l-methyl-lH- benzimidazole-5-carbonyl]-pyridin-2-yl-amino) ethyl propionate hydro bromide as one of the intermediate in order to overcome the problem of the process depicted in the product patent.

The single prodrug of Dabigatran having the formula-II,
and double

which is DabigatranEtexilate are exemplified in the examples of the patent US'380. The patent US'380 has no information about the solid state properties of the single prodrug of Dabigatran and DabigatranEtexilate. However, a similar process described in a publication of Hauel et al in Journal of Medicinal Chemistry, 2002, 45, .1757 - 1766, wherein DabigatranEtexilate is characterized by 128 - 129°C.
The PCT publication WO2006131491 discloses the anhydrous form [ of DabigatranEtexilate having the melting point 135°C, anhydrous form II of DabigatranEtexilate having the melting point 150°C, and hydrate form of DabigatranEtexilate having the melting point 90°C.
The PCT publication WO2008059029 discloses anhydrous form III of DabigatranEtexilate having melting point 128°C, anhydrous form IV of DabigatranEtexilate having the melting point 133°C, and mono hydrate form I of DabigatranEtexilate having melting point 128°C and mono hydrate form II of DabigatranEtexilate having melting point 123°C.
The different forms of the single prodrug of Dabigatran and/or the DabigatranEtexilate are disclosed in the patent applications of WO2012027543, WO2012004396 and WO 2012044595.
The patent application US2007185333 discloses the process ; for the preparation of DabigatranEtexilateMesylate from the DabigatranEtexilate by adding acetone solution of , methanesulfonic acid in an acetone solution of DabigatranEtexilate.
The patent application US 200601 83779 discloses the process for the preparation of DabigatranEtexilateMesylate from the DabigatranEtexilate by adding ethylacetate solution of methanesulfonic acid in an ethylacetate solution of DabigatranEtexilate.
Example-9: Process for the preparation of DabigatranEtexilateMesylate from DabigatranEtexilate
[0086] The DabigatranEtexilate (0.04 mol) was dissolved in acetone (250.0 ml) and added Methanesulfonic acid (0.04 mol) in Ethyl acetate (25 ml) at 25-30°C. Stirred the reaction mass for 3 hrs at the same temperature, the isolated solid was filtered and washed with acetone, dried under vacuum to get the DabigatranEtexilateMesylate. Yield: 85 %, Purity: Not less than 99.0%
Example 10: Process for the preparation of DabigatranEtexilateMesylate
[0087] To a solution of DabigatranEtexilate (0.04 mol) in Acetone (8 volumes) and Ethanol (2 volumes), Methanesulfonic acid solution [Methanesulfonic acid (0.04 mol) was dissolved in Ethyl acetate (25 ml) was added at 25-30°C and stirred for 3 hrs at the same temperature. After completion of the reaction, the resultant solid was filtered, washed with acetone and dried under vacuum. Yield: 93%
...............
l-methyl-2-|Tvi-[4-(TSi-n-hexyloxycarbonylamidino)phenyl]aminomethyl]benzimidazole- 5-yl-carboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide is commonly known as Dabigatran etexilate. Dabigatran is an anticoagulant from the class of the direct thrombin inhibitors developed by Boehringer Ingelheim and is used for the treatment of thrombosis, cardiovascular diseases, and the like. Dabigatran etexilalte mesylate was approved in both US and Europe and commercially available under the brand name Pradaxa.
Dabigatran etexilate and process for its preparation was first disclosed in WO 98/37075.
The disclosed process involves the reaction of ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2- yl)benzamido)propanoate with 2-(4-cyanophenylamino) acetic acid in the presence of N,N- carbonyldiimidazole in tetrahydrofuran to provide ethyl 3-(2-((4-cyanophenylamino)methyl)-l- methyl-N-(pyridin-2-yl)-lH-benzo[d] imidazole-5-carboxamido)propanoate, which is further converted into l-methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-ylcarboxylicacid- N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide hydrochloride by reacting with ammonium carbonate in ethanol, followed by treating with ethanolic hydrochloric acid. The obtained compound was reacted with n-hexyl chloroformate in presence of potassium carbonate in tetrahydrofuran/water provides Dabigatran etexilate and further conversion into its mesylate salt was not disclosed. The purity of Dabigatran etexilate prepared as per the disclosed process is not satisfactory, and also the said process involves chromatographic purification which is expensive and difficult to implement in the large scale. Hence the said process is not suitable for commercial scale up.
Moreover, the said process proceeds through the l-methyl-2-[N-[4-amidinophenyl] aminomethyl]benzimidazol-5-ylcarboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide hydrochloride (herein after referred as "Dabigatran hydrochloride"), which degrades to form impurities and resulting in the formation of Dabigatran etexilate with low purity. In view of intrinsic fragility of Dabigatran hydrochloride, there is a need in the art to develop a novel salt form of 1 -methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-ylcarboxylicacid-N-(2- pyridyl)-N-(2-ethoxycarbonyl ethyl)amide, which enhances the purity of the final compound.
The prior reported processes disclosed in WO2012004396 and WO2008095928 Al involves the usage of inorganic salts like hydrochloride and hydrobromide salts of ethyl 3-(2-((4- cyanophenylamino)methyl)- 1 -methyl -N-(pyridin-2-yl)- 1 H-benzo[d]imidazole-5-carboxamido) propanoate (herein after referred as "cyano intermediate") and ethyl 3-(2-((4-carbamimidoyl phenylamino)methyl)- 1 -methyl -N-(pyridin-2-yl)- 1 H-benzo[d]imidazole-5-carboxamido) propanoate (herein after referred as "amidino intermediate"). The inorganic acid addition salts are less stable when compared to the organic acid addition salts and also the process for the preparation of organic acid addition salts is very much easy when compared to inorganic acid addition salt. Inorganic acid addition salts of amidine intermediate seem to be hygroscopic in nature. Therefore, organic acid addition salts are always preferable to synthesize stable salts which in-turn enhances the purity of the final compound.
The oxalate salt of cyano intermediate was disclosed in WO2009111997. However as on date, there is no other organic acid addition salts of cyano intermediate were reported in the prior art for preparing pure Dabigatran etexilate. Henceforth, there is a need to develop a novel organic acid addition salt of cyano intermediate compound which is very much efficient when compared to its corresponding oxalate salt and that result in the formation of final compound with high purity and yield.
The process disclosed in WO 98/37075 also involves the reduction of, ethyl 3-(4- (methylamino)-3-nitro-N-(pyridin-2-yl)benzamido)propanoate (herein after referred as "nitro compound") using Pd-C in a mixture of dichloromethane and methanol under hydrogen pressure to provide ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2-yl)benzamido)propanoate (herein after referred as "diamine compound").
The reduction of nitro compound through catalytic hydrogenation in the presence of tertiary amine under hydrogen pressure was also disclosed in WO2009153214; and in presence of inorganic base under hydrogen pressure was also disclosed in WO2012004397.
However, most of the prior art processes proceed through catalytic hydrogenation which involves the pressure reactions. Handlings of these pressure reactions are not suitable for the large scale process. Therefore, there is a significant need in the art to provide a simple reduction process which avoids the difficulties associated with catalytic hydrogenation.
JMC, 2002, 45(9), 1757-1766 disclosed a process for the preparation of ethyl 3-(3-amino- 4-(methylamino)-N-(pyridin-2-yl)benzamido)propanoate starting from 4-(methylamino)-3- nitrobenzoic acid. The disclosed process involves the conversion of 4-(methylamino)-3- nitrobenzoic acid into its acid chloride using thionyl chloride and the obtained compound was reacted with ethyl 3-(pyridin-2-ylamino)propanoate to provide nitro compound, followed by catalytic reduction using Pd-C to provide diamine compound.
However, particularly in large scale synthesis the reduction reaction occasionally stops due to catalyst poisoning which leads to incomplete reaction and requires additional catalyst to complete the reaction. Moreover the sulfur impurities which are present in nitro compound formed due to the reaction with thionyl chloride in the previous stages of the synthesis of diamine compound are strongly influence the reaction time, quality and catalyst consumption in the manufacturing process.
Surprisingly, the problem associated with the catalytic hydrogenation and catalyst poisoning is solved by the present invention by adopting a suitable reducing agent such as Fe- acetic acid and Fe-hydrochloric acid.
The crystalline forms-I, II, V and VI of Dabigatran etexilate oxalate were disclosed in WO2008043759 and WO2011110876.
The crystalline forms-Ill, IV and V of Dabigatran etexilate fumarate were disclosed in WO2008043759 and WO2011110876.
Various different salts for Dabigatran etexilate and their polymorphs were reported in WO98/37075, WO03074056, WO2005028468, WO2006114415, WO2008043759, WO2011110876, WO2012027543 and WO2012044595.
The process for the preparation of crystalline form-I of Dabigatran etexilate mesylate was described in WO2005028468 and WO2012027543.
HPLC analysis of Innovator Tablet
The present inventors has also analyzed the Pradaxa 110 mg tablet having Lot no: 808809 and compared with dabigatran etexilate mesylate obtained from the present invention and found that, the impurity profile of both the products are similar to each other i.e., amide impurity, despyridyl ethyl ester etc. are well present even in Pradaxa tablet. Henceforth, we can presume that these impurities are known from the art.
Amide Impurity: 0.31%; Despyridyl ethyl ester: 0.10%; Deshexyl Impurity: 0.08%. HPLC Method of Analysis:
a) Dabigatran etexilate (Formula-1) and Dabigatran etexilate mesylate (Formula-la):
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength
UV-detector; Column: Zorbax Eclipse XDB CI 8, 100 X 4.6mm, 3.5 μιη θΓ Equivalent; Flow Rate: 1.0 mL/min; Wavelength : 300 nm; Column temperature: 25°C; Injection volume: 5 μΐ,; Run time: 50 minutes; Auto sampler temperature: 5°C; Buffer: Dissolve 0.63gm of Ammonium formate in lOOOmL of Milli-Q- Water and mix well. Adjust its pH to 8.2 with Ammonia and filtered through 0.22 μιη nylon membrane and degas it. Mobile phase-A: Buffer; Mobile phase- B: Acetonitrile: Water (80:20) v/v; Diluent: N,N-Dimethylformamide; Needle wash: Diluent; Elution: Gradient. b) Ethyl 3-(2-((4-cyanophenylamino)methyl)-l-methyl-N-(pyridin-2-yl)-lH-benzo[d] imidazole-5-carboxamido)propanoate methanesulfonate (Formula-10)
Apparatus : A liquid chromatograph is equipped with variable wavelength UV- Detector; Column: Zorbax SB CN 150 x 4.6mm, 5μπι (or) Equivalent (Make: Agilent and PNo: 883975- 905); Flow Rate: 1.0 mL / min; Column temperature: 25°C; Wave length: 290 nm; Injection volume: 5 μΐ-.; Run time: 60 minutes; Elution: Gradient; Diluent: Water: Acetonitrile (70:30) v/v; Needle wash: Diluent; Buffer: Weigh accurately about 2 g of 1 -Octane sulphonic acid sodium salt anhydrous and add 5 mL of Ortho phosphoric acid in 1000 mL of Milli-Q- Water and mix well, filter this solution through 0.22 μηι^ΐοη membrane and sonicate to degas; Mobile Phase- A: Buffer(100%);Mobile Phase- B: Acetonitrile: Methanol (90: 10) v/v. c) Ethyl 3-(2-((4-carbamimidoylphenylamino)methyl)-l-methyl-N-(pyridin-2-yl)-lH- benzo[d] imidazole-5-carboxamido)propanoate methanesulfonate (Formula-11)
Apparatus : A liquid chromatographic system is to be equipped with variable wavelength UV- Detector and Integrator; Column : Zodiac CI 8 250 X 4.6 mm, 5 μηι (or) equivalent (Make: Zodiac and PNo. ZLS.C18.46.250.0510 ); Flow Rate: 1.0 mL/min; Wavelength: 290 nm; Column temperature: 25°C; Injection Volume: 5μί; Run time: 55 min; Elution: Gradient;
Buffer: Take 5 mL of Ortho phosphoric acid(85%) and 2 g of 1 -Octane sulfonic acid sodium salt anhydrous in 1000 mL of Milli-Q-water and adjust its pH to 2.5 with Triethyl amine filter, through 0.22 μπι Nylon membrane filter paper and sonicate to degas it; Mobile Phase-A: Buffer(l 00%) Mobile Phase-B: Acetonitrile: Water (90: 10) v/v; Diluent : Water: Acetonitrile (80:20) v/v.
Morphology: Method of analysis: Samples were mounted on aluminium stubs using double adhesive tape, coated with gold using HUS-5GB vacuum evaporation and observed in Hitachi S-3000 N SEM at an acceleration voltage of 10KV.
Following are the impurities observed during the preparation of Dabigatran etexilate mesylate.

Deshexyl Impurity Despyridyl Ethyl Ester

Methyl Carbamate Ethyl Carbamate

The present invention is schematically represented as follows:

Formula-2 ene

Formula-6
Fe-AcOH

Formula-7

Dabigatran etexilate Dabigatran etexilate Mesylate The process described in the present invention was demonstrated in examples illustrated below.
Example-13: Preparation of Dabigatran etexilate (Formula-1)
n-hexanol (30.8 g) was added to a solution of N, N-carbonyldiimidazole (61.15 g) and dichloromethane (360 ml) at 15-25°C and stirred for 3 hours. The organic layer was washed with water followed by sodium chloride solution. Distilled off the solvent from the organic layer completely under reduced pressure to get amide compound. Acetonitrile (157.5 ml) was added to the obtained amide compound. This was added to a mixture of ethyl 3-(2-((4- carbamimidoylphenylamino)methyl)-l-methyl-N-( yridin-2-yl)-lH-benzo[d]imidazole-5- carboxamido)propanoate mesylate compound of formula- 11 (90 g), potassium carbonate (62.5 g), acetonitrile (378 ml) and water (252 ml) at 25-35°C. The reaction mixture was heated to 40- 50°C and stirred for 8 hours. After completion of the reaction, both the organic and aqueous layers were separated; the organic layer was cooled to -5 to +5°C and stirred for 2 hours. Filtered the precipitated solid washed with acetonitrile and water. The obtained compound was dissolved in a mixture of acetone (270 ml) and acetonitrile (270 ml) at 45-50°C. Cooled the reaction mixture to 25-30°C and water (360 ml) was added to it. Filtered the obtained solid and dissolved in the mixture of dichloromethane and sodium chloride solution at 35-40°C. Both the organic and aqueous layers were separated; the organic layer was distilled under reduced pressure and then co-distilled with ethyl acetate. The obtained crude compound was dissolved in ethyl acetate (540 ml) by heating it to 70-80°C and stirred for 30 minutes. Filtered the reaction mixture, the filtrate was cooled to 35-45°C and ethanol (8 ml) was added to the reaction mixture. The reaction mixture was again cooled to 25-35°C and stirred for 3 hours. Filtered the precipitated solid and then dried to get pure title compound.
Yield: 44 g; MR: 128-131 °C. Purity by HPLC: 99.63%.
...................




EXAMPLE 6
Preparation of dabigatran etexilate mesylate: l-methyl-2-[N-[4-( -n-hexyloxycarbonylamidino)phenyl] amino methyl]benzimidazol-5- yl-carboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonyl ethyl) amide (100 gm) was dissolved acetone (1000 ml) under heating at 25-35 °C. A solution of methane sulfonic acid (13.77 gm) in acetone (100 ml) was added to the reaction mixture. The solution is filtered and after the addition of acetone cooled to approximately 20° C. The precipitated product was filtered and washed with acetone then dried at 50° C under reduced pressure.
Wet weight : 0.120-0.140 kg
Dry weight : 0.90-1.0 kg
Yield (W/W) : 0.90-1.0
Theoretical Yield (w/w) : 1.15
Percentage Yield : 78.2-86.9%
......................
| US20050095293 * | Sep 3, 2004 | May 5, 2005 | Boehringer Ingelheim Pharma Gmbh Co. Kg | Administration form for the oral application of poorly soluble drugs |
| US20070185173 * | Dec 21, 2006 | Aug 9, 2007 | Georg Zerban | Process for the Preparation of the Salts of 4-(Benzimidazolylmethylamino)-Benzamides |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014020555A2 * | Jul 31, 2013 | Feb 6, 2014 | Alembic Pharmaceuticals Limited | An improved process for the preparation of dabigatran etexilate mesylate |
| WO2014009966A2 * | Jul 5, 2013 | Jan 16, 2014 | Rao Davuluri Ramamohan | An improved process for the preparation of dabigatran etexilate mesylate and its intermediates thereof |
| WO2014009966A3 * | Jul 5, 2013 | Mar 6, 2014 | Rao Davuluri Ramamohan | An improved process for the preparation of dabigatran etexilate mesylate and its intermediates thereof |
| EP1966171A1 | Dec 20, 2006 | Sep 10, 2008 | Boehringer Ingelheim International GmbH | Improved process for the preparation of 4-(benzimidazolylmethylamino)-benzamides and the salts thereof |
| EP1968949A1 | Dec 20, 2006 | Sep 17, 2008 | Boehringer Ingelheim International GmbH | Improved process for the preparation of the salts of 4-(benzimidazolylmethylamino)-benzamides |
| US6087380 | Feb 18, 1998 | Jul 11, 2000 | Boehringer Ingelheim Pharma Kg | Disubstituted bicyclic heterocycles, the preparations and the use thereof as pharmaceutical compositions |
| US7202368 | Jun 9, 2005 | Apr 10, 2007 | Boehringer Ingelheim International Gmbh | Process for the preparation of 4-(benzimidazolymethylamino) benzamidines |
| WO2000005207A1 * | Jul 20, 1999 | Feb 3, 2000 | Boehringer Ingelheim Pharma | Substituted phenylamidines with antithrombotic action |
| WO2007071742A1 * | Dec 20, 2006 | Jun 28, 2007 | Boehringer Ingelheim Int | Improved process for the preparation of 4-(benzimidazolylmethylamino)-benzamides and the salts thereof |
| WO2010045900A1 | Oct 26, 2009 | Apr 29, 2010 | Zentiva, K.S. | A method for the preparation of dabigatran and its intermediates |
| Reference | ||
|---|---|---|
| 1 | * | European Medicines Agency (EMEA): "CHMP ASSESSMENT REPORT FOR Pradaxa", , 1 January 2008 (2008-01-01), pages 1-36, XP55003938, London Retrieved from the Internet: URL:http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000829/WC500041062.pdf [retrieved on 2011-08-01] |
| 2 | * | HAUEL N H ET AL: "STRUCTURE-BASED DESIGN OF NOVEL POTENT NONPEPTIDE THROMBIN INHIBITORS", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 45, no. 9, 1 January 2002 (2002-01-01), pages 1757-1766, XP001098844, ISSN: 0022-2623, DOI: DOI:10.1021/JM0109513 |
| CN103058920A * | Jan 21, 2013 | Apr 24, 2013 | 上海应用技术学院 | Preparation method of 3-(2-pyridineamino)ethyl propionate |
| CN1861596A * | May 18, 2006 | Nov 15, 2006 | 复旦大学 | Process for synthesizing antithrombin inhibitor of non-asymmetric non-peptide kind |
| CN101875626A * | Nov 6, 2009 | Nov 3, 2010 | 广东光华化学厂有限公司;北京理工大学 | Method for synthesizing N-benzyl maleimide from immobilized supported acid catalyst |
| EP2522662A1 * | May 11, 2011 | Nov 14, 2012 | Medichem, S.A. | Dabigatran etexilate and related substances, processes and compositions, and use of the substances as reference standards and markers |
| JP2004315371A * | Title not available |
 WO2015124764
ERREGIERRE S.P.A. [IT/IT]; Via Francesco Baracca, 19 I-24060 San Paolo D’argon (IT)
Erregierre SpA
DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE”
Abstract
A novel process is described for the production of Dabigatran etexilate mesylate, a 5 compound having the following structural formula: and two novel intermediates of said process.
(WO2015124764) SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE click herefor patent
Dabigatran etexilate mesylate is an active substance developed by Boehringer
Ingelheim and marketed under the name Pradaxa® in the form of tablets for oral administration; Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
Dabigatran etexilate mesylate is the INN name of the compound 3-({2-[(4-{Amino-[(E)-hexyloxycarbonylimino]-methyl}-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:
WO2015124764
ERREGIERRE S.P.A. [IT/IT]; Via Francesco Baracca, 19 I-24060 San Paolo D’argon (IT)
Erregierre SpA
DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE”
Abstract
A novel process is described for the production of Dabigatran etexilate mesylate, a 5 compound having the following structural formula: and two novel intermediates of said process.
(WO2015124764) SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE click herefor patent
Dabigatran etexilate mesylate is an active substance developed by Boehringer
Ingelheim and marketed under the name Pradaxa® in the form of tablets for oral administration; Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
Dabigatran etexilate mesylate is the INN name of the compound 3-({2-[(4-{Amino-[(E)-hexyloxycarbonylimino]-methyl}-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:
 The family of compounds to which Dabigatran etexilate belongs was described for the first time in patent US 6,087,380, which also reports possible synthesis pathways.
The preparation of polymorphs of Dabigatran etexilate or Dabigatran etexilate mesylate is described in patent applications US 2006/0276513 A1 , WO 2012/027543 A1 , WO 2008/059029 A2, WO 2013/124385 A2, WO 2013/124749 A1 , WO 2013/1 1 1 163 A2 and WO 2013/144903 A1 , while patent applications WO 2012/044595 A1 , US 2006/0247278 A1 , US 2009/0042948 A2, US 2010/0087488 A1 and WO 2012/077136 A2 describe salts of these compounds.
One of the objects of the invention is to provide an alternative process for the preparation of Dabigatran etexilate mesylate and two novel intermediates of the process.
These objects are achieved with the present invention, which, in a first aspect thereof, relates to a process for the production of Dabigatran etexilate mesylate, comprising the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):
The family of compounds to which Dabigatran etexilate belongs was described for the first time in patent US 6,087,380, which also reports possible synthesis pathways.
The preparation of polymorphs of Dabigatran etexilate or Dabigatran etexilate mesylate is described in patent applications US 2006/0276513 A1 , WO 2012/027543 A1 , WO 2008/059029 A2, WO 2013/124385 A2, WO 2013/124749 A1 , WO 2013/1 1 1 163 A2 and WO 2013/144903 A1 , while patent applications WO 2012/044595 A1 , US 2006/0247278 A1 , US 2009/0042948 A2, US 2010/0087488 A1 and WO 2012/077136 A2 describe salts of these compounds.
One of the objects of the invention is to provide an alternative process for the preparation of Dabigatran etexilate mesylate and two novel intermediates of the process.
These objects are achieved with the present invention, which, in a first aspect thereof, relates to a process for the production of Dabigatran etexilate mesylate, comprising the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):
 (I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):
(I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):
 (II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):
(II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):
 (IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):
(IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):
 (VI) (VII)
e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX), or a mixture of the two compounds (VIII) and (IX):
(VI) (VII)
e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX), or a mixture of the two compounds (VIII) and (IX):
 f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):
CHsCOOH
[(VIII) ; (IX)]
f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):
CHsCOOH
[(VIII) ; (IX)]

 wherein A is a chlorine or nitrate anion;
liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,2-trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate hydrochloride (XII):
wherein A is a chlorine or nitrate anion;
liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,2-trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate hydrochloride (XII):
 reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):
reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):
 reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ‘) and isolating the latter:
reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ‘) and isolating the latter:
 j) reacting maleate salt (XI 11 ‘) with hexyl chloroformate to give compound Dabigatran etexilate (XIV :
hexyl chloroformate
j) reacting maleate salt (XI 11 ‘) with hexyl chloroformate to give compound Dabigatran etexilate (XIV :
hexyl chloroformate
 k) reacting compound (XIV) with methanesulfonic acid to give the salt Dabigatran etexilate mesylate:
k) reacting compound (XIV) with methanesulfonic acid to give the salt Dabigatran etexilate mesylate:
 a gatran etex ate mesy ate
EXAMPLE 12
Preparation of Dabigatran etexilate mesylate (step k).
All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C. Separately, a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling. The salt of the title is formed. The mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone. The precipitate is dried at 60 °C.
4.88 kg of Dabigatran etexilate mesylate, equal to 6.74 moles of compound, are obtained, with a yield in this step of 90%.
EXAMPLE 13
0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
The diffractogram shown in Figure 1 is obtained; a comparison with the XRPD data of the known Dabigatran etexilate polymorphs allows to verify that the polymorph of Example 1 1 is novel.
EXAMPLE 14
0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into
the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere. The graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.
a gatran etex ate mesy ate
EXAMPLE 12
Preparation of Dabigatran etexilate mesylate (step k).
All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C. Separately, a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling. The salt of the title is formed. The mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone. The precipitate is dried at 60 °C.
4.88 kg of Dabigatran etexilate mesylate, equal to 6.74 moles of compound, are obtained, with a yield in this step of 90%.
EXAMPLE 13
0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
The diffractogram shown in Figure 1 is obtained; a comparison with the XRPD data of the known Dabigatran etexilate polymorphs allows to verify that the polymorph of Example 1 1 is novel.
EXAMPLE 14
0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into
the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere. The graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.
 Figure 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention;
Figure 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention;
 Figure 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
1H NMR OF
Dabigatran etexilate mesylate 872728-81-9
Figure 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
1H NMR OF
Dabigatran etexilate mesylate 872728-81-9
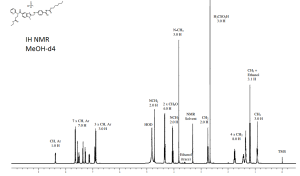 .
...
PATENT
Examples
Reference examples:
Preparation of starting material: Dabigatran etexilate mesylate form I according to US 2005/0234104 example 1:
Ethyl 3 - [(2- { [4-(hexyloxycarbonylarninoimmomemyl)phenylammo]methyl } -
1 - methyl- lH-benzimidazole-5-carbonyl)pyridm-2-ylamino]propionate base (52.6 kg) (which has preferably been purified beforehand by recrystallization from ethyl acetate) is placed in an agitator apparatus which has been rendered inert and then 293 kg of acetone is added. The contents of the apparatus are heated to 40° C to 46° C with stirring. After a clear solution has formed, the contents of the apparatus is filtered into a second agitator apparatus through a lens filter and then cooled to 30° C to 36° C. 33 kg of acetone precooled to 0° C to 5° C, 7.9 kg of 99.5% methanesulfonic acid, and for rinsing another 9 kg of acetone are placed in the suspended container of the second apparatus. The contents of the suspended container are added in metered amounts to the solution of ethyl 3-[(2-{[4-(hexyloxycarbonylamino- iminomethyl)phenylamino]methyl} - 1 -methyl- 1 H-benzimidazole-5-carbonyl)pyridin-
2- ylamino]propionate base at 26° C to 36° C within 15 to 40 minutes. Then the mixture is stirred for 40 to 60 minutes at 26° C to 33° C. It is then cooled to 17° C to 23° C and stirred for a further 40 to 80 minutes. The crystal suspension is filtered through a filter dryer and washed with a total of 270 L of acetone. The product is dried in vacuum at a maximum of 50° C for at least 4 hours. Yield: 54.5-59.4 kg;
90%-98% of theory based on ethyl 3-[(2-{[4-(hexyloxycarbonyl- ammoiminomethyl)phenylamino]methyl} - 1 -methyl- 1 H-benzimidazole-5-carbonyl)- pyridm-2-ylamino]propionate base.
Preparation of starting material: Dabigatran Etexilate free base
Dabigatran Etexilate free base can be prepared according to the procedures disclosed in US 6087380 - example 113 or US 7202368 - example 5 Example 1
2.08 g of dabigatran etexilate free base was dissolved in 14.7 ml of acetone at 30 - 36 °C. 0.210 ml of methanesulfonic acid diluted in 2.20 ml of acetone was added within 15 - 40 min. at 26 - 36 °C. The resulting mixture was first steered for 40 - 60 min. at 26 - 36 °C and then for 40 - 80 min at 17 - 23 °C.
The resulting crystalline product was filtered off, washed with 17.87 ml of acetone and dried at 50 °C for 18 hours at 540 mbar.
....................
PAPER
.
...
PATENT
Examples
Reference examples:
Preparation of starting material: Dabigatran etexilate mesylate form I according to US 2005/0234104 example 1:
Ethyl 3 - [(2- { [4-(hexyloxycarbonylarninoimmomemyl)phenylammo]methyl } -
1 - methyl- lH-benzimidazole-5-carbonyl)pyridm-2-ylamino]propionate base (52.6 kg) (which has preferably been purified beforehand by recrystallization from ethyl acetate) is placed in an agitator apparatus which has been rendered inert and then 293 kg of acetone is added. The contents of the apparatus are heated to 40° C to 46° C with stirring. After a clear solution has formed, the contents of the apparatus is filtered into a second agitator apparatus through a lens filter and then cooled to 30° C to 36° C. 33 kg of acetone precooled to 0° C to 5° C, 7.9 kg of 99.5% methanesulfonic acid, and for rinsing another 9 kg of acetone are placed in the suspended container of the second apparatus. The contents of the suspended container are added in metered amounts to the solution of ethyl 3-[(2-{[4-(hexyloxycarbonylamino- iminomethyl)phenylamino]methyl} - 1 -methyl- 1 H-benzimidazole-5-carbonyl)pyridin-
2- ylamino]propionate base at 26° C to 36° C within 15 to 40 minutes. Then the mixture is stirred for 40 to 60 minutes at 26° C to 33° C. It is then cooled to 17° C to 23° C and stirred for a further 40 to 80 minutes. The crystal suspension is filtered through a filter dryer and washed with a total of 270 L of acetone. The product is dried in vacuum at a maximum of 50° C for at least 4 hours. Yield: 54.5-59.4 kg;
90%-98% of theory based on ethyl 3-[(2-{[4-(hexyloxycarbonyl- ammoiminomethyl)phenylamino]methyl} - 1 -methyl- 1 H-benzimidazole-5-carbonyl)- pyridm-2-ylamino]propionate base.
Preparation of starting material: Dabigatran Etexilate free base
Dabigatran Etexilate free base can be prepared according to the procedures disclosed in US 6087380 - example 113 or US 7202368 - example 5 Example 1
2.08 g of dabigatran etexilate free base was dissolved in 14.7 ml of acetone at 30 - 36 °C. 0.210 ml of methanesulfonic acid diluted in 2.20 ml of acetone was added within 15 - 40 min. at 26 - 36 °C. The resulting mixture was first steered for 40 - 60 min. at 26 - 36 °C and then for 40 - 80 min at 17 - 23 °C.
The resulting crystalline product was filtered off, washed with 17.87 ml of acetone and dried at 50 °C for 18 hours at 540 mbar.
....................
PAPER
 .
.| Chinese Journal of Applied Chemistry |
Synthesis of Dabigatran Etexilate
|
| LIU Xiaojun, CHEN Guohua* |
| (Department of Medicinal Chemistry,China Pharmaceutical University,Nanjing 210009,China) |
4-Methylamino-3-nitrobenzoic acid(3) was prepared from 3-nitro-4-chlorobenzoic acid by methylamination. 3-[(Pyridin-2-yl)amino]propinoic acid ethyl ester(5) was prepared from 2-aminopyridine and ethyl acrylate by Michael addition. Dabigatran etexilate was synthesized from compounds 3 and 5 via condensation, catalytic hydrogenation, acylation with N-(4-cyanophenyl)glycine(9), cyclization, Pinner reaction, followed by reaction with n-hexyl chlorofomate. The overall yield is about 40% and the structure of the product was determined by IR, 1H NMR and MS.
ESIMS(m/z):628[M+H]+;1 HNMR(500MHz,DMSOd6), δ:091(t,3H,J=90Hz,CH3),116(t,3H,J=85Hz,CH3),125~169(m,8H,4×CH2),274(t, 2H,J=145Hz,CH2CO),379(s,3H,CH3N),395~403(m,4H,2CH2O),428(t,2H,J=140Hz, CH2),451(d,2H,J=55Hz,CH2N),676(d,J=85Hz,2H,Ar—H),688(d,J=75Hz,1H,Ar— H), 702(s,1H,N—H),713~721(m,2H,Py—H),740(d,J=85Hz,1H,Ar—H),747(d,J=15Hz, 1H,Ar—H),755~759(m,1H,Py—H),786(d,J=85Hz,2H,Ar—H),836~843(m,1H,Py—H), 902(brs,2H,NH2);IR(KBr),σ/cm-1 :3374,2953,2929,1730,1640,1610,1470,1389,1326,1256,1192, 1145,1127,1021,945,835,811,768,747。



............
PAPER
Identification, Synthesis, and Strategy for the Reduction of Potential Impurities Observed in Dabigatran Etexilate Mesylate Processes
† Novel Technology Center of Pharmaceutical Chemistry, Shanghai Institute of Pharmaceutical Industry, 1111 North Zhongshan No. 1 Road, Shanghai 200437, P. R. China
‡ Department of Pharmacy, Shandong Provincial Hospital affiliated to Shandong University, Jinan 250021,P. R. China
Org. Process Res. Dev., 2014, 18 (6), pp 744–750
DOI: 10.1021/op500084q
 Synthetic impurities that are present in dabigatran etexilate mesylate were studied, and possible pathways by which these impurities are formed during the manufacturing process were examined. The impurities were monitored by high-performance liquid chromatography, and their structures were determined by mass spectrometry and 1H and 13C NMR. Potential causes for the formation of these impurities are discussed, and strategies to minimize their formation are also described.
Synthetic impurities that are present in dabigatran etexilate mesylate were studied, and possible pathways by which these impurities are formed during the manufacturing process were examined. The impurities were monitored by high-performance liquid chromatography, and their structures were determined by mass spectrometry and 1H and 13C NMR. Potential causes for the formation of these impurities are discussed, and strategies to minimize their formation are also described.
 ................
1H NMR PREDICT
................
1H NMR PREDICT

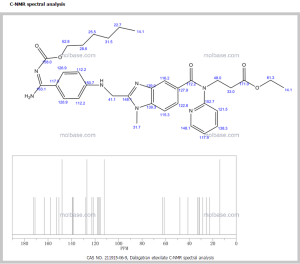 13 C NMR PREDICT ABOVE
13 C NMR PREDICT ABOVE
† Novel Technology Center of Pharmaceutical Chemistry, Shanghai Institute of Pharmaceutical Industry, 1111 North Zhongshan No. 1 Road, Shanghai 200437, P. R. China
‡ Department of Pharmacy, Shandong Provincial Hospital affiliated to Shandong University, Jinan 250021,P. R. China
Org. Process Res. Dev., 2014, 18 (6), pp 744–750
DOI: 10.1021/op500084q


| WO2015044375A1 * | Sep 26, 2014 | Apr 2, 2015 | Ratiopharm Gmbh | Pharmaceutical preparation comprising dabigatran etexilate bismesylate |
See full gatran series at..................http://apisynthesisint.blogspot.in/p/argatroban.html
3
ARGATROBAN
ArgatrobanMolecular Formula: C23H36N6O5S
Formula Weight: 508.63
CAS No.: 74863-84-6
Formula Weight: 508.63
CAS No.: 74863-84-6
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
PATENT
US 7,589,106, 7,687,516, EP 0008746; US 4258192, US 4201863
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).

Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.
……………………………………………………….
Argatroban monohydrate

Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl) amino]-1-oxo2[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2- piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. cas 141396-28-3
Argipidine, Argatroban monohydrate, GN1600, DK-7419, MDI-805 Acova, Slonnon, Novastan
Mitsubishi Chemical (Originator), Encysive Pharmaceuticals (Licensee), Mitsubishi Pharma (Distributor), Daiichi Pharmaceutical (Codevelopment), GlaxoSmithKline (Codevelopment), Mitsubishi Pharma (Codevelopment), Sanofi-SynthLabo (Codevelopment)
Antithrombocytopenic, CARDIOVASCULAR DRUGS, Cerebrovascular Diseases, Treatment of, HEMATOLOGIC DRUGS, Hematopoiesis Disorders Therapy, Ischemic Stroke, Treatment of, NEUROLOGIC DRUGS, Peripheral Vascular Disease, Treatment of, Stroke, Treatment of, Treatment of Peripheral Obstructive Vascular Disease, Thrombin Inhibitors
Synthesis of argatroban on the method reported in the literature there are two synthetic routes, patent EP8746, US4258192, US4201863, JP8115267 relates to a route is: with 4 – methyl-piperidine as a starting material was prepared first intermediate body (2R, 4R) -4 – methyl-2 – ethyl-piperidine, and the first and a t-BOC protected amino nitro-L-arginine condensation, and then the 3 – methyl – 8 – quinoline sulfonyl chloride condensation after hydrolysis, hydrogenation, hydration be argatroban. This entry route synthesis process complicated procedure to be carried out under the protection of nitrogen, the raw material is highly toxic gas phosgene, the operation more difficult.
US4117127, JP02-212473, EP823430, EP8746, JC S Perk Transl 1981 (5), JP02-212473 relates to an alternative route is: nitro L-arginine prior to the 3 – methyl-8 – subsequent condensation quinoline sulfonyl chloride Intermediate (2R, 4R) -4 – methyl-2 – piperidinecarboxylate condensation, and then after hydrolysis, hydrogenation, hydration be argatroban. This synthetic route despite the relatively simple process method, to obtain raw materials, but this method using reagents such as phosphorus oxychloride, phosphorus trichloride has a pungent odor, easy to absorb moisture in humid air, intense smoke, environmental pollution, greater stimulation of the body’s respiratory tract, can cause eye and skin irritation and burning, and the use of this method, complex operation, low yield, high cost.
Patent CN100586946C Argatroban discloses a method for separating optically active isomeric compounds, the feedstock argatroban mixed solvent of alcohol and water was heated to reflux 5-10 hours, cooled and allowed to stand, and filtered to give White crystalline product, dried, repeated 2-6 times. [0008] Patent CN101033223A discloses a Argatroban is the main by-product (2R, 4R)-l_ [N2-(3_ methyl-8 – quinolinesulfonyl)-L-arginyl] -4 – methyl-2 – carboxylic acid, argatroban, and the byproducts are difficult to isolate, argatroban two diastereomeric isomer 21 (S) and 21 (R) separation of work attracted a lot of research persons. Because both physical and chemical properties are very similar, so separation is very difficult. 1993 Rawson, Thomas E.; VanGorp, Kimmie A.; Yang, Janet so first by high pressure liquid chromatography and column chromatography separation to obtain a single 21 (S) and 21 (R) argatroban [ Journal of Pharmaceutical Sciences vol. 82, No. 6,672]; Thibaudeau Karen et al. reported Protein A chromatographic separation [US6440417]. However, due to the separation of these methods a small amount of low efficiency, so there is no practical value industrialization. 2006 China Tianjin Weijie Technology Co., Ltd. Song Honghai et al. Reported using recrystallization Separation 21 (S) and 21 (R) argatroban way [0 With 951,936 it], so that the mass 21 (5) Aga music classes as possible, but the law of low yield, complicated operation, high cost, and a large amount of a small amount of 21 (S) of 21 (R)-product argatroban, from the viewpoint of industrial production, is still a ideal method. [0009] These methods can be effectively prepared argatroban, but the purity of the desired product is not high, poor color, content is low, affecting the quality of the results of its preparation.
U.S. Pat. No. 4,201,863 (6 May 1980) and EP 8746 (filed on 22 Aug. 1979 with priority based on the application for the cited US patent) describe a class of N2-arylsulphonyl-L-argininamide drugs, with anti-thrombotic activity, and the processes for obtaining them. Of these, the compound 4-methyl-1-[N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (argatroban, isomers mixture) is described. The described process comprises the synthesis of an intermediate NG-substituted-N2-quinolinesulphonyl-L-argininamide from which the desired compound is obtained by catalyzed hydrogenolysis or acidolysis and catalyzed hydrogenation. The general conditions provided for the hydrogenolysis and hydrogenation reaction are: i) inert solvents (methanol, ethanol, tetrahydrofuran or dioxane); ii) presence of a catalyst (Raney nickel, palladium, platinum, ruthenium, rhodium); iii) hydrogen atmosphere at a pressure between 1 and 100 kg/cm2 and preferably between 5 and 50 kg/cm2; iv) temperature between 0° C. and 200° C. and preferably between 50° C. and 150° C.; v) reaction temperature from 2 hours to 120 hours. The crude product obtained is then purified by trituration or by re-crystallization from diethyl ether-tetrahydrofuran, diethyl ether-methanol or from water-methanol or by chromatography. No example is given of this purification step. In particular, both U.S. Pat. No. 4,210,863 and EP 8746 in example 1(E) describe the preparation of argatroban, isomers mixture. This compound is obtained in amorphous form by hydrogenation of [NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid in ethanol in the presence of Pd/C with hydrogen pressure of 10 kg/cm2 at 100° C. for 8 hours. The catalyst is removed by filtration of the ethanol solution which is then evaporated without further purification and/or re-crystallization steps. In the US patent at issue as indeed in patent application EP 8746, no mention is made of polymorphic forms of the compounds and, for the obtained compound, the following characteristics are reported: Amorphous solid, I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.01; H 6.98; N 16.61.
U.S. Pat. No. 4,258,192 (24 Mar. 1981) (continuation-in-part of the aforesaid patent application U.S. Pat. No. 4,201,863) and the same patent application EP 8746 describe the stereoisomers and the preparation thereof, including argatroban used as an active principle in medicaments, i.e. the stereoisomer (2R,4R)-4methyl-[4N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid, with the following characteristics: melting point (m.p.). 188-191° C.; I.R. (KBr) (cm−1) 3400, 1620, 1460, 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.05; H 6.94; N 16.65. The compound is prepared according to the description given in examples 1(E) in U.S. Pat. No. 4,258,192 and 2(E) and 3 in EP 8746 respectively by hydrogenation of (2R,4R) 1-[NG-nitro-N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid in ethanol in presence of acetic acid catalyzed by Pd/C. After filtering the mass to remove the catalyst, the solvent is evaporated and the residue suspended in chloroform, the solution treated with a saturated sodium bicarbonate solution or 1N sodium hydroxide solution and after washing, the solvent is evaporated. The compound is then re-crystallized from ethanol. Again in this case, no reference is made to the obtainment of monohydrate polymorphic forms.
Said polymorphic forms are described instead in the publication Biochem. Biophys. Res. Comm. 1981, 101, 440-446 in the context of stereoisomer preparation. The monohydrate polymorph of the (2R,4R) stereoisomer is prepared by re-crystallization from ethanol/water and the reported characteristics are: m.p. 176-180° C.; [α]D 27 +76.1° (c 1, 0.2N HCl).
U.S. Pat. No. 5,925,760 (20 Jul. 1999) and EP 0823430 (filed 4 Aug. 1997) subsequently describe a new method for preparing argatroban by means of a new intermediate N2-(3-methyl-8-quinolinesulphonyl)-NG-nitro-L-arginine. In particular the patent makes reference to the preparation of a crystalline monohydrate form of argatroban, referring back to examples (D) and (E) of Japanese patent publication No. (Hei)-2-31055/1990 and generically to an I.R. spectrum identical to that of the commercially available argatroban compound. The relevant example in the cited patent publication is example (E), while example (D) concerns the preparation of (2R,4R)-1-[NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid. This compound represents the starting compound for argatroban preparation by catalytic reduction in the presence of Pd/C. The crude argatroban obtained is then purified by extraction with chloroform, treatment with a saturated sodium bicarbonate solution and, after solvent evaporation, re-crystallization from ethanol or from 15% alcohol in water. It should be noted however that the Japanese patent makes no mention of the monohydrate form of argatroban being obtained and that for the compound the following characteristics are reported: m.p. 188-191° C.; molecular composition (theoretical/found) (%): C 54.31/54.01; H 7.13/6.98; N 16.52/16.61; I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380. These analytical data, with the exception of the unreported melting point, are the same as those indicated in the cited patent documents describing a mixture of (2R,4R)-4methyl-[4N2-(3S-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid and (2R,4R)-4methyl-1-[N2-(3R-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid isomers of argatroban, but do not correspond to the melting point given in the publication, being the only document that identifies the monohydrate form of argatroban.
More recently, patent application CN 1,951,937 (filing date 10 Nov. 2006) described a method for preparing hydrated argatroban by treating argatroban with large quantities of water (more than 60 and up to 80 volumes of distilled water per gram of argatroban) at a temperature of 80-100° C. for a time of 0.5-1 hour and crystallization by cooling. The water content reported is comprised between 3.3 and 3.8% and the ratio of dextroisomer R to levoisomer S is R:S=63-67: 37-33.
Argatroban is a compound of wide therapeutic use, for which reason the need still exists to provide a compound of pharmaceutically acceptable quality obtained by easily industrialized and economically convenient methods. With regard to the monohydrate, this form is preferable for the applicative purpose since the anhydrous form is unstable and tends to become hydrated and/or wet. Moreover it crystallizes only with difficulty at the correct ratio between the diastereoisomers.

…..

the protection of 4-methylpiperidine (I) with (Boc)2O gives the carbamate (II), which is condensed with benzyl chloroformate by means of sec-butyl lithium and TMEDA in ethyl ether to yield (?-trans-1-(tert-butoxycarbonyl)-4-methylpiperidine-2-carboxylic acid benzyl ester (III). Deprotection of the NH group of (III) with HCl in ethyl acetate affords (?-trans-4-methylpiperidine-2-carboxylic acid benzyl ester (IV), which is condensed with the protected arginine derivative (V) by means of isobutyl chloroformate and TEA to provide the corresponding amide as a diastereomeric mixture. Resolution of this mixture by flash chromatography furnishes the desired diastereomer (VI), which is treated with HCl in ethyl acetate in order to remove the Boc-protecting group to yield compound (VII). Condensation of compound (VII) with 3-methylquinoline-8-sulfonyl chloride (VIII) by means of TEA in dichloromethane affords the expected sulfonamide (IX). Finally, this compound is submitted to hydrogenation with H2 over Pd/C in AcOH/ethanol in order to produce debenzylation, cleavage of the NO2 group and hydrogenation of the pyridine ring to yield argatroban.
………….
Argatroban, i.e., (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino) pentyl)-4-methyl-2-piperidine carboxylic acid, has two diastereoisomers: 21(R) and 21(S). Usually the ratio of 21(R) to 21(S) is 64-65: 36-35 (U.S. Pat. No. 6,440,417, Cossy. J., et al, Bioorganic & Medicine Chemistry Letters, 11 (2001), 1989-1992, Journal of pharmaceutical Sciences, Vol. 82, No. 6, 672 (1993)).
The structure formula of Argatroban is reported below:

- 21(S) Argatroban, X=CH3, Y═H;
- 21(R) Argatroban, X=H, Y=CH3;
- Argatroban, 21(S): 21 (R)=35:65.
The chemical names of the two diastereoisomers mentioned above are:
- 21(S) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3S)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-72-6); and
- 21(R) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3R)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-71-5).
In 1978, S. Akamoto et al from Japanese Mitsubishi Chemical Corporation first disclosed the anti-thrombin activity of Argatroban monohydrate (U.S. Pat. No. 4,101,653). In the next 20 years, numerous researchers had in-depth studies on Argatroban about its biological activity and medicine values. In 1981, S. Akamoto compared Argatroban with heparin in vivo (Okamoto, S. et al., Biochem. Biophys. Res. Commun. 101, 440 (1981)); T. Kumoto disclosed its three-dimensional selective activity (Kumada, T. et al., Thromb. Res. 24, 285 (1981)). In 1984, R. Kumato made a clinical evaluation of hemodialysis of Argatroban (Kikumoto, R. et al., Biochemistry 23, 85 (1984)), and in 1986, he further disclosed that Argatroban can inhibit the thrombin activity of mammals, and can be used as active ingredient to treat and prevent thrombosis and as an inhibitor of platelet aggregation. Argatroban monohydrate can be used as a selective anti-thrombosis agent for treatment of chronic arterial blockage and cerebral thrombosis, etc (JP 61-48829). In 1992 and 1993, Taparelli and Jakubowski separately disclosed the reversibility of Argatroban in anti-thrombin (Taparelli, C., Trends Pharmacol. Sci., 1993, 14, 366, Jakubowski, J. A. et al, Rep. Med. Chem., 1992, 27, 99). In 1990s, many researchers such as L. R. Buch reported other related research (Buch, L. R., Cadiosvasc. Drug Rev., 1991, 9, 247, Strupcnewski, J. D. et al., Academic: San Diego, 1991; Vol. 26, p 299, Brundish, D. et al., J. Med. Chem. 1999; 42, 4584, Shebuski, R. J., Academic: San Diego, 1999; Vol. 26, p 98). In 1992, Argatroban monohydrate was first approved as an anti-thrombin medicine in Japan (Hijikata-Okunomiya, A., et al, Thromb. Hemostasis, 1992, 18, 135).
Updated in sept 2015, reader there may be duplication
| Argatroban |
| AC1L99H9; AC-15185; TL8005144; C04931; | |
| MOLECULAR FORMULA: | C23H36N6O5S |
|---|---|
| MOLECULAR WEIGHT: | 508.63414 g/mol |
(2R,4R)-1-[5-(diaminomethylideneamino)-2-[(3-methyl-1,2,3,4-tetrahydroquinolin-8-yl)sulfonylamino]pentanoyl]-4-methylpiperidine-2-carboxylic acid, cas 74863-84-6
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Prodrugs of (2R)-2-Propyloctanoic Acid For the Treatment of Stroke [US7495029] | 2008-06-05 | 2009-02-24 |
Tomiya Mano, Jin Shiomura, “Argatroban preparations for ophthalmic use.” U.S. Patent US5506241, issued October, 1986.
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in theliver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).
Argatroban is a direct, selective thrombin inhibitor. The American College of Cardiologists (ACC) recommend using bivalirudin or argatroban in patients who have had, or at risk for, heparin induced thrombocytopenia (HIT) and are undergoing percutaneous coronary intervention. Argatroban is a non-heparin anticoagulant shown to both normalize platelet count in patients with HIT and prevent the formation of thrombi. Parental anticoagulants must be stopped and a baseline activated partial thromboplastin time must be obtained prior to administering argatroban.

Transitioning to warfarin in individuals with heparin induced thrombocytopenia
Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.

http://www.google.com/patents/WO2009124906A2?cl=en
………….
NMR paper
Complete 1H and 13C assignments of (21R) and (21S) diastereomers of argatroban
Article first published online: 20 DEC 2007
DOI: 10.1002/mrc.2122, http://onlinelibrary.wiley.com/doi/10.1002/mrc.2122/abstract



click on image for clear view
1H NMR PREDICT


13C NMR PREDICT


References
1 Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005;353:1028-40. PMID 16148288
3Dhillon S. Argatroban: A Review of its Use in the Management of Heparin-Induced Thrombocytopenia. Am J Cardiovasc Drugs 2009; 9 (4): 261-82. Link text
- Hursting MJ, Lewis BE, Macfarlane DE. (2005). “Transitioning from argatroban to warfarin therapy in patients with heparin-induced thrombocytopenia.”. Clin Appl Thromb Hemost 11 (3): 279–87. doi:10.1177/107602960501100306. PMID 16015413.
External links
| EP0823430A1 * | Aug 4, 1997 | Feb 11, 1998 | Mitsubishi Chemical Corporation | Method for preparing n2-arylsulfonyl-l-argininamides |
| US4201863 * | Aug 31, 1978 | May 6, 1980 | Mitsubishi Chemical Industries, Limited | N2 -Arylsulfonyl-L-argininamides and the pharmaceutically acceptable salts thereof |
| REFERENCE | ||||
|---|---|---|---|---|
| 1 | None | |||
| 2 | * | OKAMOTO S ET AL: “Potent inhibition of thrombin by the newly synthesized arginine derivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 101, no. 2, 30 July 1981 (1981-07-30), pages 440-446, XP024844713 ISSN: 0006-291X [retrieved on 1981-07-30] cited in the application | ||
| 3 | * | SONG H: “Method for preparing argatroban monohydrate in pure water” CASREACT,, 25 April 2007 (2007-04-25), XP002493272 & CN 1 951 937 A (TIANJIN WEIJIE TECHNOLOGY CO L [CN]) 25 April 2007 (2007-04-25) | ||
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
| WO2012136504A1 | Mar 26, 2012 | Oct 11, 2012 | Lundbeck Pharmaceuticals Italy S.P.A. | Method for the preparation of process intermediates for the synthesis of argatroban monohydrate |
| CN102408468A * | Sep 20, 2011 | Apr 11, 2012 | 海南灵康制药有限公司 | Argatroban compound and preparation method thereof |
| EP2752412A1 | Mar 26, 2012 | Jul 9, 2014 | Lundbeck Pharmaceuticals Italy S.p.A. | Intermediates for the synthesis of Argatroban monohydrate |
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5[(aminoiminomethyl)amino]1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
|
Argatroban Injection is a sterile, non-pyrogenic, clear, colorless to pale yellow isotonic solution. It is supplied in a single use polyolefin bag containing 250 mg of argatroban in 250 mL sodium chloride solution (1 mg/mL). Each mL contains 1 mg argatroban, 9 mg sodium chloride, USP, and 3 mg sorbitol, NF in water for injection, USP. The pH of the solution is between 3.2 to 7.5.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2- [[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl] sulfonylamino]pentanoyl]-4-methyl-piperidine-2- carboxylic acid | |
| CLINICAL DATA | |
| TRADE NAMES | Argatroban |
| AHFS/DRUGS.COM | monograph |
| ROUTES OF ADMINISTRATION | intravenous |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | 100% (intravenous) |
| PROTEIN BINDING | 54% |
| METABOLISM | hepatic |
| BIOLOGICAL HALF-LIFE | 39 and 51 minutes |
| IDENTIFIERS | |
| CAS REGISTRY NUMBER | 74863-84-6 |
| ATC CODE | B01AE03 |
| PUBCHEM | CID: 440542 |
| DRUGBANK | DB00278 |
| CHEMSPIDER | 389444 |
| UNII | OCY3U280Y3 |
| KEGG | C04931 |
| CHEMBL | CHEMBL1166 |
| CHEMICAL DATA | |
| FORMULA | C23H36N6O5S |
| MOLECULAR MASS | 508.635 g/mol |
//////
4
//////////
This comment has been removed by the author.
ReplyDeleteNice informative post..
ReplyDeletepayroll software
visitor management software
mobile app development