







BOROLE COMPDS
1 AN 3485
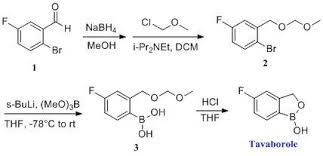
2 TAVABOROLE
3 GSK 2251052, Epetraborole, AN3365
4 Crisaborole, AN 2728
5 AN-2718
6 AN 2898
7
8
9
10
WILL BE UPDATED...........
1 AN 3485

AN 3485

AN3485,
6-(4-(aminomethyl)-2-chlorophenoxyl)benzo[c][1,2]oxaborol-1(3H)-ol,
was synthesized at Anacor Pharmaceuticals as described in patent application WO 2010028005
A1
6-[4-(Aminomethyl)-2-chlorophenoxy]-2,1-benzoxaborol-1(3H)-ol hydrochloride
Anacor Pharmaceuticals, Inc.
http://jpet.aspetjournals.org/content/early/2012/11/28/jpet.112.200030.full.pdf
Pro-inflammatory cytokines play a critical role in the development of autoimmune and
inflammatory diseases. Targeting the cytokine environment has proven efficient for averting
inflammation. In this study, we reported that 6-(4-(aminomethyl)-2-
chlorophenoxyl)benzo[c][1,2]oxaborol-1(3H)-ol (AN3485), a benzoxaborole analog, inhibited
TLR2-, TLR3-, TLR4- and TLR5-mediated TNF-α, IL-1β and IL-6 release from human PBMCs
and isolated monocytes with IC50s ranging from 18 to 580 nM, and the inhibition was mediated
at the transcriptional level. Topical administration of AN3485 significantly reduced PMAinduced contact dermatitis and oxazolone-induced delayed-type hypersensitivity in mice,
indicating its capability of penetrating skin and potential topical application in skin
inflammation. Oral administration of AN3485 showed dose-dependent suppression of LPSinduced TNF-α and IL-6 production in mice with an ED90 of 30 mg/kg. Oral AN3485, 35
mg/kg, twice a day, suppressed collagen-induced arthritis in mice over a 20-day period. The
potent anti-inflammatory activity in in vitro and in vivo disease models makes AN3485 an
attractive therapeutic lead for a variety of cutaneous and systemic inflammatory diseases
A new class of boron-containing small molecules has been developed over the past several
years as potential drugs. Different from carbon, boron contains an electrophilic empty p-orbital
which can form transient bonds with nucleophiles in an enzyme active site, which mimics a
tetrahedral transition state of peptide bond cleavage in an enzymatic reaction (Baker et al., 2011).
The benzoxaboroles, in which the boron atom is incorporated into a heteroaromatic ring system,
are able to inhibit a number of important enzymes, including bacterial and fungi Leucyl-tRNA
synthetase (Rock et al., 2007), human phosphodiesterase-4 (PDE4) (Akama et al., 2009) and
HCV NS3/4A protease (Li et al., 2010). Three benzoxaboroles, AN2690 (Tavaborole), AN2728
and AN3365 (GSK’052) are in clinical trials for treatment of onychomycosis, psoriasis/atopic dermatitis and Gram-negative bacterial infection, and have been proven safe in human when
applied topically or systemically
……………………………………………………….
Structure-activity relationships of 6-(aminomethylphenoxy)-benzoxaborole derivatives as anti-inflammatory agent
Bioorg Med Chem Lett 2013, 23(6): 1680
http://www.sciencedirect.com/science/article/pii/S0960894X13001054

Scheme 2.
Synthesis of compounds 9a–e. Reagents and conditions: (a) K2CO3,
DMSO, 80–90 °C, overnight (33–61%); (b) LAH, THF, 0 °C to rt, 1 h, then
4 M HCl in 1,4-dioxane (43–68%); (c) aq NaOH, MeOH, 50 °C, 2 h (61%),
(d) Ac2O, pyridine, rt (79%).
Patent
https://www.google.com/patents/WO2010028005A1?cl=en

Compound 2:
To a solution of 2-hydroxy-4-methoxy-benzaldehyde (30 g, 197 mmol) in DCM (anhydrous, 120 rnL) was added pyridine (79 mL, 986 mmol) at room temperature. After the mixture was cooled to -10 0C, the Tf2O (50 mL, 296 mmol) was slowly added to the reaction between -10 0C to 0 0C. The addition took about 2.5 hours. After the addition, the stirring was kept for 30 minutes. The EtOAc (200 mL) was added. The organic layer was washed with 1 M HCl (3 X 80 mL), dried over MgSO4, filtered, and evaporated under vacuum. The residue was purified over silica gel, eluting with 5% EtOAc / hexanes to give trifluoro-methanesulfonic acid 2- formyl-5-methoxy-phenyl ester (2) 46 g in 82% yield. 1H NMR (400 MHz,
CHLOROFORM-^) δ ppm 10.13 (s, 1 H), 7.95 (d, J=8.99 Hz, 1 H), 7.03 (dd, J=8.60, 2.34 Hz, 1 H), 6.88 (d, J=2.34 Hz, 1 H), 3.93 (s, 3 H)
Compound 3:
To a solution of trifluoro-methanesulfonic acid 2-formyl-5-methoxy-phenyl ester (2) (46 g, 160 mmol) in 1,4-dioxane (anhydrous, 360 rnL) were added bis(pinacolato)diboron (82.3 g, 320 mmol), [l,l ‘-bis(diphenylphosphino)ferrocene] palladium(II)chloride (23.7 g, 32 mmol) and KOAc (47.6 g, 480 mmol). The mixture was stirred at room temperature with N2bubbling for 30 minutes. Then the reaction was heated at 100 0C for 3 hours. The solution was filtered, evaporated under vacuum. The residue was purified over silica gel, eluting with 20% EtOAc / hexanes to afford 4-methoxy-2-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzaldehyde (3) 37.8 g in 90% yield. 1H NMR (400 MHz, CHLOROFORM- d) δ ppm 10.34 (s, 1 H), 7.90 (d, J=8.60 Hz, 1 H), 7.26 (s, 1 H), 6.99 (d, J=8.60 Hz, 1 H), 3.86 (s, 3 H), 1.36 (s, 12 H)
Compound 4:
To a clear solution of 4-methoxy-2-(4,4,5,5-tetramethyl-
[l,3,2]dioxaborolan-2-yl)-benzaldehyde (3) (48 g, 180 mmol) in MeOH (anhydrous, 300 mL) was slowly added NaBH4 (6.96 g, 180 mmol). The reaction was stirred at room temperature for 2 hours. Then IM HCl (100 mL) was slowly added. After stirring for overnight, the MeOH was evaporated under vacuum. The solid was filtered, washed with water and air-dried to afford 6-methoxy-3H- benzo[c][l,2]oxaborol-l-ol (4) 23 g in 77% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.11 (s, 1 H), 7.29 (d, J=8.21 Hz, 1 H), 7.23 (d, J=2.34 Hz, 1 H), 7.03 (dd, J=8.40, 2.54 Hz, 1 H), 4.90 (s, 2 H), 3.75 (s, 3 H).
Compound 5:
To a clear solution of 6-methoxy-3H-benzo[c][l,2]oxaborol-l-ol (4) (600 mg, 3.66 mmol) in DCM (anhydrous, 60 mL) was slowly added BBr3 (1M/DCM, 8.05 mL, 8.05 mmol) at -10 0C. The reaction was stirred for 3 hours, with monitoring by NMR. After all 4 had gone, 30 mL of cold water was added. Then 50 mL of EtOAc was added to extract all organic compounds. The organic layer was washed with cold brine, until the pH of aqueous layer changed to pH 7. The organic layer was dried over Na2SO4, filtered, evaporated under vacuum. The residue (-85% HPLC purity) was used directly for the next step reaction without further purification. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.29 (s, 1 H), 9.04 (s, 1 H), 7.17 (d, J=8.21 Hz, 1 H), 7.07 (d, J=2.34 Hz, 1 H), 6.85 (dd, J=8.21, 2.34 Hz, 1 H), 4.85 (s, 2 H). ESMS (m/z): 149 (M- H)“. HPLC: 88.31% (220 nm), 85.02% (maxplot).
Compound 6:
To a solution of 3H-benzo[c][l,2]oxaborole-l,6-diol (5) (300 mg, 2 mmol) in DMSO (30 mL) were added K2CO3 (828 mg, 6 mmol) and 3-chloro-4-fiuoro- benzonitrile (933 mg, 6 mmol). The reaction was heated at 90 0C for 7 hours. After the cooling of reaction solution, EtOAc (50 mL) was added. The organic layer was washed with water (5 X 50 mL). The organic layer was evaporated under vacuum. The residue was purified by reverse phase chromatography to afford 3-chloro-4-(l- hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yloxy)-benzonitrile (6) 190 mg in 33.3% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.24 (s, 1 H), 8.22 (s, 1 H), 7.77 (d, J=7.81 Hz, 1 H), 7.50 (d, J=8.20 Hz, 1 H), 7.34 (s, 1 H), 7.28 (d, J=8.20 Hz, 1 H), 7.01 (d, J=8.59 Hz, 1 H), 4.99 (s, 2 H). ESMS (m/z): 284 (M-H)“. HPLC: 96.41% (220 nm), 96.0% (maxplot).
(X): IS AN 3485
To a clear solution of 3-chloro-4-(l-hydroxy-l,3-dihydro- benzo[c][l,2]oxaborol-6-yloxy)-benzonitrile (6) (136 mg, 0.48 mmol) in THF
(anhydrous, 60 mL) was added lithium aluminum hydride (lM/ether, 1.19 mL, 1.19 mmol) at 0 0C. The reaction was stirred for 2 hours. Then the reaction was quenched with IM HCl (30 mL). MeOH (50 mL) was added and the solution was filtered. The filtrate was evaporated under vacuum. The residue was purified by reverse phase chromatography (biotage, gradient MeOH / H2O from 10% to 100%) to afford (X) 106 mg (white solid) in 68% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.19 (s, 1 H), 8.18 (br, s, 3 H), 7.75 (s, IH), 7.44-7.39 (m, 2 H), 7.19-7.10 (m, 3 H), 4.98 (s, 2 H), 4.03 (q, J=5.50 Hz, 2 H).
ESMS (m/z): 290 (M+H)+.
HPLC: 95.9% (220 nm), 96.85% (maxplot).
.
2
TAVABOROLE
TAVABOROLE
- AN 2690
- AN-2690
- AN2690
- UNII-K124A4EUQ3
5-Fluoro-2,1-benzoxaborol-1(3H)-ol;
1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole
MOLECULAR FORMULA C7H6BFO2
MOLECULAR WEIGHT 151.9
SPONSOR Anacor Pharmaceuticals, Inc.
CAS REGISTRY NUMBER 174671-46-6
Mp 118-120° C.....US20070265226
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H)
FDA APPROVED JULY 2 2014..........."FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails". Market Watch. July 8, 2014.
PHASE 3
Has antifungal activity.
The US Food and Drug Administration (FDA) 2014 JULY 8 ratified the Anacor's Kerydin (5% Tavaborole solution) for the topical treatment of nail fungal infections. Tavaboroleindications of toenail fungus Trichophyton rubrum or Trichophyton rubrum infections.Instructions recommended once a day for toenail infections, treatment for 48 weeks, on the recommendation of Anacor, and do not need to nail debridement.
I tis an oxaborole antifungal used topically, as a 5% w/w solution, for the treatment of onychomycosis of the toenails due to Trichophyton rubrumor T. mentagrophytes. It is applied to the affected toenail once daily for 48 weeks.
Ingrowing toenails and application site reactions including exfoliation, erythema, and dermatitis have been reported during use.

1H NMR FROM NET

CLICK ON IMAGE FOR CLEAR VIEW
COSY NMR PREDICT

Tavaborole (AN2690, trade name Kerydin) is a topical antifungal medication for the treatment of onychomycosis, a fungal infectionof the nail and nail bed. Tavaborole began its Phase 3 trials in December 2010[1] and was approved in July 2014.[2] Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection. No treatment-related systemic side effects were observed in any of its clinical trials.
Tavaborole is the first oxygen boron used to treat toenail infections dioxolane (oxaborole) antifungal agents, located in Palo Alto, Anacor focuses on boron-based drug development and production, according to the latest news, Tavaborole future also be used to infect fingernails. Wedbush Securities analyst predicts that next year the drug sales in the United States for $ 16 million, by 2021 will reach peak sales of $ 347 million.
Gram-negative bacteria cause approximately 70% of the infections in intensive care units. A growing number of bacterial isolates responsible for these infections are resistant to currently available antibiotics and to many in development. Most agents under development are modifications of existing drug classes, which only partially overcome existing resistance mechanisms. Therefore, new classes of Gram-negative antibacterials with truly novel modes of action are needed to circumvent these existing resistance mechanisms. We have previously identified a new a way to inhibit an aminoacyl-tRNA synthetase, leucyl-tRNA synthetase (LeuRS), in fungi via the oxaborole tRNA trapping (OBORT) mechanism.
Herein, we show how we have modified the OBORT mechanism using a structure-guided approach to develop a new boron-based antibiotic class, the benzoxaboroles, which inhibit bacterial leucyl-tRNA synthetase and have activity against Gram-negative bacteria by largely evading the main efflux mechanisms in Escherichia coli and Pseudomonas aeruginosa. The lead analogue, is active against Gram-negative bacteria, including Enterobacteriaceaebearing NDM-1 and KPC carbapenemases, as well as P. aeruginosa. This novel boron-based antibacterial, has good mouse pharmacokinetics and was efficacious against E. coli and P. aeruginosa in murine thigh infection models, which suggest that this novel class of antibacterials has the potential to address this unmet medical need.
Anacor continued development on that drug, tavaborole, and filed for FDA approval in July. The FDA will review the phase 3 trial data and issue a decision on July 29, 2014.
If approved, Anacor hopes tavaborole's ability to clear onychomycosis in 10% of treated patients will be enough to win market share away from generic Lamisil and generic topical Pentac. While Lamisil cleared the fungus in 38% of patients, it's been associated with rare cases of liver failure. And Pentac requires frequent debridement of the nail and only clears the fungus in 5.5% to 8.5% of patients.
Tavaborole is a novel, topical antifungal medication being developed for the topical treatment of onychomycosis, a nail fungus infection, which affects seven to ten percent of the U.S. population. Early studies show AN-2690 penetrates the nail effectively and has robust activity against dermatophytes, which cause onychomycosis.


1H NMR PREDICT



.......................................................................................
13 C NMR PREDICT


ARTICLE

July 18, 2013
Abstract Accepted for Oral Presentation at the 2013 American Podiatric Medical Association Annual Scientific Meeting
PALO ALTO, Calif.--(BUSINESS WIRE)-- Anacor Pharmaceuticals (NASDAQ:ANAC) announced today that its abstract "Pivotal Phase 3 Safety and Efficacy Results of Tavaborole (Formerly AN2690), a Novel Boron-Based Molecule for the Topical Treatment of Toenail Onychomycosis" was accepted for oral presentation at the 2013 APMA Annual Scientific Meeting (The National) to be held in Las Vegas, Nevada. Max Weisfeld, DPM, will present the data from tavaborole's Phase 3 studies on Monday, July 22, 2013 during the Evidence-Based Medicine and Oral Abstracts session.
As announced earlier this year, tavaborole achieved statistically significant and clinically meaningful results on all primary and secondary endpoints in two Phase 3 pivotal studies without concomitant debridement. Anacor is seeking approval for tavaborole from the Food and Drug Administration (FDA) and will file a New Drug Application imminently. Currently, there is only one FDA-approved topical treatment for onychomycosis, a fungal infection of the nail and nail bed, which affects approximately 35 million people in the United States.

"I'm impressed with tavaborole's safety and efficacy data. There is no FDA-approved topical treatment for onychomycosis with tavaborole's range of efficacy and ability to penetrate the nail to reach the site of the infection," said Dr. Weisfeld. "Tavaborole's Phase 3 results demonstrate its ability to clear the nail and eliminate the infection which is important to both patients and the physicians who treat them. In addition, tavaborole is easy to apply and dries quickly which makes it convenient for patients to use."
"We are pleased to present these positive data at the APMA's Annual Scientific Meeting, the leading annual meeting of podiatrists. As we seek FDAapproval for tavaborole, we look forward to developing relationships with podiatrists to potentially offer them a new treatment option for the large number of patients who seek treatment for onychomycosis," said David Perry, Chief Executive Officer of Anacor Pharmaceuticals.
About the Studies
Anacor conducted two separate Phase 3 studies of tavaborole on patients with distal subungual onychomycosis affecting 20 to 60 percent of the target great toenail. Approximately 600 patients aged 18 years and older with no upper age limit (the oldest subject was 88 years old) were enrolled in each study and randomized two-to-one to receive either tavaborole or the vehicle control. Patients were instructed to apply tavaborole solution or the vehicle to the toenail once daily for 48 weeks.
A copy of the presentation will be available on Anacor's website following the oral session.
About Anacor Pharmaceuticals
Anacor is a biopharmaceutical company focused on discovering, developing and commercializing novel small-molecule therapeutics derived from its boron chemistry platform. Anacor has discovered eight compounds that are currently in development. Its two lead product candidates are topically administered dermatologic compounds — tavaborole, a topical antifungal for the treatment of onychomycosis, and AN2728, a topical anti-inflammatory PDE-4 inhibitor for the treatment of atopic dermatitis and psoriasis. In addition to its two lead programs, Anacor has discovered three other wholly-owned clinical product candidates — AN2718 and AN2898, which are backup compounds to tavaborole and AN2728, respectively, and AN3365 an antibiotic for the treatment of infections caused by Gram-negative bacteria. We have discovered three other compounds that we have out-licensed for further development — two compounds for the treatment of animal health indications that are licensed to Eli Lilly and Company and AN5568, also referred to as SCYX-7158, for human African trypanosomiasis (HAT, or sleeping sickness), which is licensed to Drugs for Neglected Diseases initiative, or DNDi. We also have a pipeline of other internally discovered topical and systemic boron-based compounds in development. For more information, visit http://www.anacor.com.
.................................................................................................
Patents
WO 1995033754
WO 2004009578....
WO 2006089067
WO 2008025543
................................
SYNTHESIS
Drugs Fut 2006, 31(8): 667
J Med Chem 2006, 49(15): 4447
.................................
Reference:
ELI LILLY AND COMPANY Patent: WO2004/9578 A2, 2004 ; Location in patent: Page 36-37 ; WO 2004/009578 A2...........................
PATENT
Anacor Pharmaceuticals Patent: US2007/265226 A1, 2007 ; Location in patent: Page/Page column 59 ;
http://www.google.com/patents/US20070265226

1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole (19b)
To
a solution of 5b (73.2 g, 293 mmol) in dry THF (400 mL) was added
n-butyllithium (1.6 M in hexanes; 200 mL) over 45 min at −78° C. under
nitrogen atmosphere. Anion precipitated. After 5 min, (i-PrO)3B
(76.0 mL, 330 mmol) was added over 10 min, and the mixture was allowed
to warm to room temperature over 1.5 h. Water and 6 N HCl (55 mL) were
added, and the solvent was removed under reduced pressure to about a
half volume. The mixture was poured into ethyl acetate and water. The
organic layer was washed with brine and dried over anhydrous Na2SO4.
The solvent was removed under reduced pressure. To a solution of the
residue in tetrahydrofuran (360 mL) was added 6 N HCl (90 mL), and the
mixture was stirred at 30° C. overnight. The solvent was removed under
reduced pressure to about a half volume. The mixture was poured into
ethyl acetate and water. The organic layer was washed with brine and
dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was treated with i-Pr2O/hexane to give 19b (26.9 g, 60%) as a white powder:
mp 118-120° C.; 1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H);
ESI-MS m/z 151 (M−H)−;
HPLC purity 97.8%; Anal (C7H6BFO2) C, H.
.....................
Gunasekera, Dinara S.; Gerold, Dennis J.; Aalderks, Nathan S.; Chandra, J. Subash; Maanu, Christiana A.; Kiprof, Paul; Zhdankin, Viktor V.; Reddy, M. Venkat Ram Tetrahedron, 2007 , vol. 63, # 38 p. 9401 - 9405
.........................
Baker, Stephen J.; Zhang, Yong-Kang; Akama, Tsutomu; Lau, Agnes; Zhou, Huchen; Hernandez, Vincent; Mao, Weimin; Alley; Sanders, Virginia; Plattner, Jacob J. Journal of Medicinal Chemistry, 2006 , vol. 49, # 15 p. 4447 - 4450
....................
Ding, Charles Z.; Zhang, Yong-Kang; Li, Xianfeng; Liu, Yang; Zhang, Suoming; Zhou, Yasheen; Plattner, Jacob J.; Baker, Stephen J.; Liu, Liang; Duan, Maosheng; Jarvest, Richard L.; Ji, Jingjing; Kazmierski, Wieslaw M.; Tallant, Matthew D.; Wright, Lois L.; Smith, Gary K.; Crosby, Renae M.; Wang, Amy A.; Ni, Zhi-Jie; Zou, Wuxin; Wright, Jon Bioorganic and Medicinal Chemistry Letters, 2010 , vol. 20, # 24 p. 7317 - 7322
..............
PATENT
US20050261277
PREPARATION 13 5-Fluoro-3H-benzo[c][1,2)oxaborol-1-ol

Dissolve 1-bromo-2-(1-ethoxy-ethoxymethyl)-4-fluoro-benzene(5.4 g, 19.5 mmol) in dry THF (100 mL) and cool to −78° C. under nitrogen. Add butyl lithium (2.5M in Hexanes, 10.2 mL, 25.4 mmol) dropwise at −78° C. Upon complete addition, stir the reaction at −78° C. for 10 minutes and then add trimethyl borate (4.4 mL, 39 mmol) and warm the reaction to room temperature. Pour the reaction into 1N HCl (100 mL) and stir for 1 hour. Extract the biphasic mixture with ether three times. Dry the combined organic layers with sodium sulfate, filter and concentrate in vacuo. Triturate the oily residue with cold hexanes to yield 2.1 g (70%) of the title compoud as a white solid.
1H NMR (d6-DMSO)
9.18 (s, 1H),
7.70 (dd, J=8.2, 5.8 Hz, 1H),
7.20 (dd, J=9.5, 2.7 Hz, 1H),
7.11 (m, 1H), 4.92 (s, 1H).
.....................
SEE
http://jpet.aspetjournals.org/content/early/2012/11/28/jpet.112.200030.full.pdf
......................................
SEE
Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis.
Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ.
J Med Chem. 2006 Jul 27;49(15):4447-50.
Boron-containing inhibitors of synthetases.
J Med Chem. 2006 Jul 27;49(15):4447-50.
Boron-containing inhibitors of synthetases.
Baker SJ, Tomsho JW, Benkovic SJ.
Chem Soc Rev. 2011 Aug;40(8):4279-85. doi: 10.1039/c0cs00131g. Epub 2011 Feb 7. Review.
Chem Soc Rev. 2011 Aug;40(8):4279-85. doi: 10.1039/c0cs00131g. Epub 2011 Feb 7. Review.
- Benzoxaborole
antimalarial agents. Part 2: Discovery of fluoro-substituted
7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles.Zhang YK, Plattner JJ, Freund YR, Easom EE, Zhou Y, Ye L, Zhou H, Waterson D, Gamo FJ, Sanz LM, Ge M, Li Z, Li L, Wang H, Cui H.
Bioorg Med Chem Lett. 2012 Feb 1;22(3):1299-307. doi: 10.1016/j.bmcl.2011.12.096. Epub 2011 Dec 28.
Tavaborole Market Opportunity
Anacor is developing tavaborole specifically to address the current limitations of existing treatment options for onychomycosis. This includes designed leaps forward in both the potential safety and efficacy profile aimed to make the drug a best-in-class therapy. Additionally, management has used the company's expertise in medicinal chemistry to improve delivery of the compound through the nail plate to the nail bed, the site of onychomycosis infection. For example, preclinical studies indicate that tavaborole is able to penetrate the nail plate 250 times more effectively than ciclopirox.
Tavaborole novel mechanism of action inhibits an essential fungal enzyme, leucyl transfer RNA synthetase, or LeuRS required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection.
Likewise, the topical dosing was designed to eliminate systemic absorption. Previous preclinical and clinical data shows topical treatment with tavaborole resulted in little or no detectable levels of drug in the blood or urine. No treatment related systemic side effects have been observed in any clinical trials to date. Safety data from the company's studies to date was recently presented at the 100th National APMA meeting in Washington, DC.
Anacor's topical solution currently in two phase III trials for onychomycosis. Phase II data with tavaborole suggests efficacy superior to ciclopirox with little to no systemic exposure.
Data from an open-label phase 2 program with tavaborole showed 50% patients using a 7.5% solution saw 2 mm clear nail growth and negative fungal cultures after six months. Roughly 25% of the patients saw 5 mm clear nail growth and negative fungal cultures after six months.I see onychomycosis as a significant market opportunity for Anacor. An estimated 35 million Americans have nail fungus, with about 95% of the infections in the toenail. With efficacy similar to Lamisil, we think Anacor can capture 20% of the market. With a price per course of treatment at around $1,200, I think peak sales of tavaborole are $500 million.
Anacor and partner Merck (NYSE:MRK) met with the U.S. FDA in 2009 to discuss the phase II data. Merck has since returned the rights to tavaborole to Anacor. The original deal was with Schering-Plough in 2007. Merck most likely felt as though tavaborole clashed with existing products or did not have peak sales potential large enough to continue the partnership with Anacor. We see tavaborole as a specialty promoted product, into podiatrists and dermatologists. For a company like Anacor, it's an attractive first product.
Anacor's first phase III trial completed enrollment in November 2011. The second phase III trial completed enrollment in December 2011. Data from these trials are expected around the middle of January 2013. Data from the second study is expected six weeks later. Given the positive phase II data noted above, we think odds favor a positive outcome. A benchmark for the trial is the efficacy of Lamisil, which is a complete cure rate of around 35% to 40%, and a mycological cure of around 70% after a typical course of treatment.
I note that on Anacor's third quarter conference call management noted that they are pleased with the conduct of the trial to date. Specifically, the compliance rate appears to better than management had expected. The trial was designed with a 20% drop-out rate. It looks as though the drop-out rate is only around 13%, at a minimum suggestive of good safety and tolerability, but potentially also a sign that the drug is working.
Conclusion
I'll note two more important pieces of information for investors. Firstly, besides optimism for tavaborole, Anacor has apipeline of anti-infectant drugs. For this article I discussed only tavaborole. A second article can be dedicated entirely to AN2728 for the treatment of psoriasis and atopic dermatitis. Anacor also has an animal health collaboration with Eli Lilly (NYSE:LLY).
The second important thing to note is Anacor's cash position. The company reported financial results on November 7, 2012. The company held $36.6 million in cash on the balance sheet as of September 30, 2012. However, in October 2012, the company completed an underwritten public offering of 4.0 million shares of common stock at $6.00 per share to raise net proceeds of $22.7 million. I view the current cash position as sufficient to report data from both phase 3 trials and, if positive, file the new drug application (NDA) around the middle of 2013.
With phase 3 data expected in less than two months, good prior evidence of both safety and efficacy, and a solid cash position, I think Anacor could be an attractive investment at today's price. The stock is down meaningfully over the past month and investors can buy sizably below the October offering.
SYNTHESIS

References
- Clinical trial number NCT01270971 at ClinicalTrials.gov
- "FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails". Market Watch. July 8, 2014.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204427s000lbl.pdf
- http://www.molbase.com/en/hnmr_174671-46-6-moldata-1568017.html#tabs
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
5-Fluoro-2,1-benzoxaborol-1(3H)-ol
| |
| Clinical data | |
| Trade names | Kerydin |
| Legal status |
|
| Routes of administration | Topical use only |
| Identifiers | |
| CAS Registry Number | 174671-46-6 |
| ATC code | None |
| PubChem | CID: 11499245 |
| ChemSpider | 9674047 |
| Synonyms | AN2690 |
| Chemical data | |
| Formula | C7H6BFO2 |
| Molecular mass | 151.93 g/mol |
NMR PREDICT
H-NMR spectral analysis
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole H-NMR spectral analysis |

C-NMR spectral analysis
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole C-NMR spectral analysis |

more

 Anacor
Anacor
BIG TEAM Hernandez, front row, fifth from left, poses during a research meeting at Naeja’s headquarters.
Anacor Pharmaceuticals
is out to change that. The Palo Alto, Calif.-based biotechnology
company is developing a family of boron-containing small-molecule drugs.
And with the assistance of Naeja Pharmaceutical,
a Canadian contract research organization, Anacor has licensed one of
those molecules to GlaxoSmithKline and taken another one into Phase III
clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets...........https://pubs.acs.org/cen/coverstory/89/8912cover3.html
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets...........https://pubs.acs.org/cen/coverstory/89/8912cover3.html
UPDATED.............
http://www.apexbt.com/downloader/document/A3177/NMR.pdf
mp 118-120....http://www.syninnova.com/catalog/product/SL-264
http://www.apexbt.com/downloader/document/A3177/HPLC.pdf
antifugal AN2690 by Anacor
Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis.
Minimum Inhibitory Concentration: 1, 1, 0.5, 0.25, and 0.25 μg/mL for T.rubrum, T.mentagrophytes, C.albicans, C.neoformans, A.fumigatus, respectivley.
AN2690 is a new boron-containing antifungal agent for the potential treatment of onychomycosis. Onychomycosis is caused mainly by dermatophytes, a class of fungus that dwells on skin, hair, and nails and is the cause of other cutaneous fungal infections such as athlete’s foot.
In vitro: AN2690 showed the most active against fungi and especially against the dermatophytes T. rubrum and T. mentagrophytes, the primary fungal pathogens causing onychomycosis. In addition, AN2690 was identified as having a unique profile of in vitro antidermatophyte activity, maintenance of this activity in the presence of keratin, and exceedingly good penetration of human nails [1].
Ex vivo: AN2690 was found to have superior penetration compared to ciclopirox, and achieves levels within and under the nail plate that suggest it has the potential to be an effective topical treatment for onychomycosis [2].
Clinical trial: The efficacy of tavaborole as a topical treatment for onychomycosis has been evaluated in two identical randomised, double-blind phase III studies, NCT01270971 (301) and NCT01302119 (302), enrolling 593 and 601 patients, respectively. Completely or almost clear nail and negative mycology was achieved in 15.3 and 17.9 % of tavaborole recipients compared with 1.5 and 3.9 % of vehicle recipients [3]
References:
[1] Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49(15):4447-50.
[2] Hui X, Baker SJ, Wester RC, Barbadillo S, Cashmore AK, Sanders V, Hold KM, Akama T, Zhang YK, Plattner JJ, Maibach HI. In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96(10):2622-31.
[3] Markham A. Tavaborole: first global approval. Drugs. 2014;74(13):1555-8.
mp 118-120....http://www.syninnova.com/catalog/product/SL-264
http://www.apexbt.com/downloader/document/A3177/HPLC.pdf
antifugal AN2690 by Anacor
Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis.
Minimum Inhibitory Concentration: 1, 1, 0.5, 0.25, and 0.25 μg/mL for T.rubrum, T.mentagrophytes, C.albicans, C.neoformans, A.fumigatus, respectivley.
AN2690 is a new boron-containing antifungal agent for the potential treatment of onychomycosis. Onychomycosis is caused mainly by dermatophytes, a class of fungus that dwells on skin, hair, and nails and is the cause of other cutaneous fungal infections such as athlete’s foot.
In vitro: AN2690 showed the most active against fungi and especially against the dermatophytes T. rubrum and T. mentagrophytes, the primary fungal pathogens causing onychomycosis. In addition, AN2690 was identified as having a unique profile of in vitro antidermatophyte activity, maintenance of this activity in the presence of keratin, and exceedingly good penetration of human nails [1].
Ex vivo: AN2690 was found to have superior penetration compared to ciclopirox, and achieves levels within and under the nail plate that suggest it has the potential to be an effective topical treatment for onychomycosis [2].
Clinical trial: The efficacy of tavaborole as a topical treatment for onychomycosis has been evaluated in two identical randomised, double-blind phase III studies, NCT01270971 (301) and NCT01302119 (302), enrolling 593 and 601 patients, respectively. Completely or almost clear nail and negative mycology was achieved in 15.3 and 17.9 % of tavaborole recipients compared with 1.5 and 3.9 % of vehicle recipients [3]
References:
[1] Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49(15):4447-50.
[2] Hui X, Baker SJ, Wester RC, Barbadillo S, Cashmore AK, Sanders V, Hold KM, Akama T, Zhang YK, Plattner JJ, Maibach HI. In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96(10):2622-31.
[3] Markham A. Tavaborole: first global approval. Drugs. 2014;74(13):1555-8.
3 GSK2251052, Epetraborole, AN3365,

(S)-3-(Aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy-1,3-dihydro- 2,1-benzoxaborole (GSK2251052) is a novel boron-containing antibiotic that inhibits bacterial leucyl tRNA synthetase, and that has been in development for the treatment of serious Gramnegative infections
(S)-3-aminomethylbenzoxaborole; ABX; AN-3365; GSK ‘052; GSK-052; GSK-2251052, GSK2251052, Epetraborole
[(S)-3-(aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy- 1,3-dihydro-2,1-benzoxaborole hydrochloride],
(S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride
1-Propanol, 3-(((3S)-3-(aminomethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaborol-7-yl)oxy)-
AN3365,MW 237.0614,
cas 1093643-37-8
UNII: 6MC93Z2DF9
Anacor Pharmaceuticals, Inc., INNOVATOR
Glaxosmithkline Llc DEVELOPER
Biomedical Advanced Research and Development Authority (BARDA)
GSK 2251052 • $38.5M over the 1st two years; up to $94M……..http://www.idsociety.org/uploadedFiles/IDSA/Policy_and_Advocacy/Current_Topics_and_Issues/Advancing_Product_Research_and_Development/Bad_Bugs_No_Drugs/Press_Releases/FIS%20Slides.pdf

Originally came from Anacor about ten years ago, then was picked up by GlaxoSmithKline, and it’s an oxaborole heterocycle that inhibits leucyl tRNA synthetase
GlaxoSmithKline recently announced a contract with the Biomedical Advanced Research and Development
Authority (BARDA), a US government preparedness organization , The award guarantees GSK $38.5 million over 2 years towards development of GSK2251052, a molecule co-developed with Anacor Pharma a few years back, as a counter-bioterrorism agent. The full funding amount may later increase to $94 million, pending BARDA’s future option.
The goal here is to develop “GSK ‘052”, as it’s nicknamed among med-chemists, into a new antibiotic against especially vicious and virulent Gram negative bacteria, such as the classic foes plague (Yersinia pestis) or anthrax (Bacillus anthracis).
Look closely at GSK’052 (shown above): that’s a boron heterocycle there! Anacor, a company specializing in boron based lead compounds, first partnered with GSK in 2007 to develop novel benzoxaborole scaffolds. This isn’t the first company to try the boron approach to target proteins; Myogenics (which, after several acquisitions, became Millennium Pharma) first synthesizedbortezomib, a boronic acid peptide, in 1995.


Stephen Benkovic (a former Anacor scientific board member) and coworkers at Penn State first discovered Anacor’s early boron lead molecules in 2001, with a screening assay. The molecules bust bacteria by inhibiting leucyl-tRNA synthetase, an enzyme that helps bacterial cells to correctly tag tRNA with the amino acid leucine. Compounds with cyclic boronic acids “stick” to one end of the tRNA, rendering the tRNA unable to cycle through the enzyme’s editing domain. As a result, mislabeled tRNAs pile up, eventually killing the bacterial cell.
Inhibition of synthetase function turns out to be a useful mechanism to conquer all sorts of diseases. Similar benzoxaborozoles to GSK ‘052 show activity against sleeping sickness (see Trypanosoma post by fellow Haystack contributor Aaron Rowe), malaria, and various fungi.
Boron-containing molecules such as benzoxaboroles that are useful as antimicrobials have been described previously, see e.g. “Benzoxaboroles – Old compounds with new applications” Adamczyk-Wozniak, A. et al., Journal of Organometallic Chemistry Volume 694, Issue 22, 15 October 2009, Pages 3533-3541 , and U.S. Pat. Pubs. US20060234981 and US20070155699. Generally speaking, a benzoxaborole has the following structure and substituent numbering system:

Epetraborole R-mandelate
1234563-15-5
Epetraborole hydrochloride
1234563-16-6
Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets.
 Lucy
Shapiro is a Professor in the Department of Developmental Biology at
Stanford University School of Medicine where she holds the Virginia and
D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman
Center for Molecular and Genetic Medicine. She is a member of the
Scientific Advisory Board of Ludwig Institute for Cancer Research and is
a member of the Board of Directors of Pacific Biosciences, Inc. She
founded the anti-infectives discovery company, Anacor Pharmaceuticals,
and is a member of the Anacor Board of Directors. Professor Shapiro has
been the recipient of multiple honors, including: election to the
American Academy of Arts and Sciences, the US National Academy of
Sciences, the US Institute of Medicine, the American Academy of
Microbiology, and the American Philosophical Society. She was awarded
the FASEB Excellence in Science Award, the 2005 Selman Waksman Award
from the National Academy of Sciences, the Canadian International 2009
Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime
Achievement Award, the 2012 Horwitz Prize and President Obama awarded
her the National Medal of Science in 2012. Her studies of the control of
the bacterial cell cycle and the establishment of cell fate has yielded
valuable paradigms for understanding the bacterial cell as an
integrated system in which the transcriptional circuitry is interwoven
with the three-dimensional deployment of key regulatory and
morphological proteins, adding a spatial dimension to the systems
biology of regulatory networks.
Lucy
Shapiro is a Professor in the Department of Developmental Biology at
Stanford University School of Medicine where she holds the Virginia and
D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman
Center for Molecular and Genetic Medicine. She is a member of the
Scientific Advisory Board of Ludwig Institute for Cancer Research and is
a member of the Board of Directors of Pacific Biosciences, Inc. She
founded the anti-infectives discovery company, Anacor Pharmaceuticals,
and is a member of the Anacor Board of Directors. Professor Shapiro has
been the recipient of multiple honors, including: election to the
American Academy of Arts and Sciences, the US National Academy of
Sciences, the US Institute of Medicine, the American Academy of
Microbiology, and the American Philosophical Society. She was awarded
the FASEB Excellence in Science Award, the 2005 Selman Waksman Award
from the National Academy of Sciences, the Canadian International 2009
Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime
Achievement Award, the 2012 Horwitz Prize and President Obama awarded
her the National Medal of Science in 2012. Her studies of the control of
the bacterial cell cycle and the establishment of cell fate has yielded
valuable paradigms for understanding the bacterial cell as an
integrated system in which the transcriptional circuitry is interwoven
with the three-dimensional deployment of key regulatory and
morphological proteins, adding a spatial dimension to the systems
biology of regulatory networks.Stephen J. Benkovic
- Evan Pugh University Professor and Eberly Chair in Chemistry
414 Wartik Laboratory
University Park, PA 16802
Email: sjb1@psu.edu
(814) 865-2882
http://chem.psu.edu/directory/sjb1
Websites,Benkovic Research Group
Naeja was a three-year-old contract research firm run by Ronald Micetich and his son Christopher Micetich. Based in Edmonton, Alberta, the firm is staffed by chemists and biologists from a variety of nations who have found Canada welcoming to highly educated immigrants.
GSK. Last July, the British firm paid Anacor $15 million and exercised its option to take over development of AN3365. David J. Payne, vice president of GSK’s antibacterial drug discovery unit, lauded the compound, now renamed GSK2251052, as having “the potential to be the first new-class antibacterial to treat serious hospital gram-negative infections in 30 years.” GSK chemists have since developed a stereospecific synthesis for commercial-scale production
David J. Payne, vice president of GSK’s antibacterial drug discovery unit
David J Payne Dr Payne holds a BSc in Biochemistry from Brunel University, UK, and a PhD and DSc from The Medical School, University of Edinburgh, UK. Dr Payne has 20 years of experience in antibacterial drug discovery and is currently Vice President and Head of the Antibacterial Discovery Performance Unit (DPU) within the Infectious Diseases Centre of Excellence in Drug Discovery (ID CEDD) where he is responsible for GSK’s antibacterial research effort from discovery to clinical proof of concept (up to Phase II clinical trials). At GSK, Dr Payne has played a leading role in redesigning the strategy for antibacterial research and has helped create long-term alliances with innovative biotechnology companies which has expanded GSK’s discovery pipeline. Furthermore, he has created industry-leading partnerships with the Wellcome Trust and the Defense Threat Reduction Agency (US Department of Defense) to accelerate GSK’s antibacterial programmes. To date, Dr Payne has been involved with the progression of a broad diversity of novel mechanism antibacterial agents into development. Dr Payne has authored more than 190 papers and conference presentations.
PATENT
http://www.google.com/patents/US20090227541
- General procedure for Chiral Synthesis of 3-aminomethylbenzoxaboroles
-


- 4-(3-Aminomethyl-1-hydroxy-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyramide acetate salt (A5)

-

-
A mixture of 4-(2-bromo-3-formyl-phenoxy)-butyric acid ethyl ester (5.50 g, 17.5 mmol), bis(neopentyl glucolato)diboron (6.80 g, 30.1 mmol), PdCl2(dppf).CH2Cl2 (1.30 g, 1.79 mmol), and KOAc (5.30 g, 54.1 mmol) in anhydrous THF (600 mL) was heated with stirring at 80° C. (bath temp) O/N under an atmosphere of N2. The mixture was then filtered through Celite and concentrated in vacuo to approximately one quarter of the original volume. The resulting precipitate was isolated by filtration. The precipitate was washed with THF and EtOAc and the combined filtrate was concentrated in vacuo to give an oily residue which was used directly in the next reaction without further purification.
-
1H NMR (400 MHz, CDCl3) δ (ppm): 9.95 (s, 1H), 7.47-7.39 (m, 2H), 7.09-7.07 (m, 1H), 4.14 (q, J=7.2 Hz, 2H), 4.09-4.01 (m, 2H), 3.83 (s, 3H), 3.66 (s, 3H), 2.53 (t, J=8.0 Hz, 2H), 2.19-2.07 (m, 2H), 1.32-1.22 (m, 3H), 0.98 (s, 6H).
-

-
MeNO2 (1.3 mL, 25 mmol) was added dropwise to a stirred solution of crude 4-[2-(5,5-dimethyl-[1,3,2]dioxaborinan-2-yl)-3-formyl-phenoxy]-butyric acid methyl ester (9.4 g), NaOH (1.0 g, 25 mmol) and H2O (35 mL) in MeCN (90 mL) at rt. The mixture was stirred at rt O/N and then acidified (pH 2) using 4 M HCl. The THF was removed in vacuo and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (10% to 30% EtOAc in hexane) to give the title compound as a yellow oil: yield 2.52 g (45% over 2 steps).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.04 (s, 1H), 7.46-7.42 (m, 1H), 7.07-7.05 (m, 1H), 6.88-6.86 (m, 1H), 5.87 (d, J=8.2 Hz, 1H), 5.69 (dd, J=9.2, 2.5 Hz, 1H), 5.29 (dd, J=13.3, 2.7 Hz, 1H), 4.14-3.94 (m, 5H), 2.55-2.44 (m, 2H), 2.02-1.88 (m, 2H), 1.16 (t, J=7.2 Hz, 3H); MS (ESI) m/z=322 (M−1, negative).
-

-
A mixture of 4-(1-hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid ethyl ester (2.51 g, 7.78 mmol), 10% NaOH (17 mL), and 1:1 MeOH/H2O (70 mL) was stirred at rt for 5 h. The MeOH was removed in vacuo and the remaining aqueous layer was acidified to pH 1 using 2 M HCl. The aqueous layer was then extracted with EtOAc. The organic fractions were washed with brine, dried (MgSO4), and concentrated in vacuo to give the title compound as a pale yellow foam: yield 1.85 g (81%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.08 (bs, 1H), 9.01 (bs, 1H), 7.46-7.41 (m, 1H), 7.06-7.04 (m, 1H) 6.89-6.87 (m, 1H), 5.70 (dd, J=7.0, 2.3 Hz, 1H), 5.30 (dd, J=13.3, 2.3 Hz, 1H), 4.55 (dd, J=13.6, 4.2 Hz, 1H), 4.03 (t, J=6.6 Hz, 2H), 2.40 (t, J=7.5 Hz, 2H), 1.95-1.89 (m, 2H); MS (ESI) m/z=296 (M+1, positive).
- 3-Aminomethyl-6-(2-hydroxy-propoxy)-3H-Benzo[c][1,2]oxaborol-1-ol acetate salt (A31)

-

-
A mixture of 2-bromo-4-fluoro-benzaldehyde (30.0 g, 148 mmol), Na2CO3 (78.31 g, 738.8 mmol) and 2-benzyloxy propanol (24.56 g, 147.8 mmol) in anhydrous DMSO (300 mL) was heated with stirring at 130° C. (bath temp) for 72 h under N2. The reaction mixture was cooled to rt and diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O then brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (hexane to 30% EtOAc in hexane) to give the title compound: yield 3.84 g (7%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.22 (s, 1H), 7.88 (d, J=8.6 Hz, 1H), 7.42-7.20 (m, 5H), 7.12 (d, J=2.3 Hz, 1H), 6.92 (dd, J=8.8, 2.2 Hz, 1H), 4.52 (s, 2H), 4.16 (t, J=6.2 Hz, 2H), 3.65 (t, J=6.1 Hz, 2H), 2.10 (q, J=6.2 Hz, 2H).
-

-
General procedure 5: 4-(2-benzyloxy propoxy-2-bromobenzaldehyde (4.84 g, 13.9 mmol), B2pin2 (5.27 g, 20.8 mmol), KOAc (4.08 g, 41.6 mmol), PdCl2(dppf).CH2Cl2 (811 mg, 8 mol %), and 1,4-dioxane (50 mL). Purification: Biotage (gradient from 2% EtOAc/hexane to 20% EtOAc/hexane): yield 4.0 g (70%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.36 (s, 1H), 7.93 (d, J=8.6 Hz, 1H), 7.43-7.14 (m, 6H), 7.01 (dd, J=8.6, 2.7 Hz, 1H), 4.53 (s, 2H), 4.18 (t, J=6.2 Hz, 2H), 3.66 (t, J=6.1 Hz, 2H), 2.11 (q, J=6.1 Hz, 2H), 1.40 (s, 12H).
-

-
General procedure 8: 4-(2-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (3.0 g, 7.6 mmol), MeNO2 (924 mg, 15.1 mmol), NaOH (605 mg, 15.1 mmol), and H2O (10 mL). Purification: flash chromatography (10% EtOAc/hexane to 40% EtOAc): yield 820 mg (30%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.46 (bs, 1H), 7.45 (d, J=8.2 Hz, 1H), 7.41-7.18 (m, 6H), 7.09 (dd, J=8.6, 2.3 Hz, 1H), 5.71 (dd, J=9.2, 2.5 Hz, 1H), 5.31 (dd, J=13.3, 2.7 Hz, 1H), 4.58-4.40 (m, 3H), 4.08 (t, J=6.2 Hz, 2H), 3.60 (t, J=6.2 Hz, 2H), 2.08-1.94 (m, 2H).
-

-
General procedure 13: 6-(2-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (820 mg, 2.29 mmol), 20% Pd(OH)2 (850 mg, 1 equiv w/w), and AcOH (40 mL). Purification: preparative HPLC: yield 120 mg (22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.32 (d, J=8.2 Hz, 1H), 7.22 (s, 1H), 7.02 (d, J=7.8 Hz, 1H), 4.98 (bs, 1H), 4.04 (t, J=6.2 Hz, 2H), 3.56 (t, J=6.2 Hz, 2H), 3.03-2.85 (m, 1H), 2.61 (dd, J=12.9, 7.0 Hz, 1H), 1.89 (s, 3H), 1.97-1.67 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 97.44% (MaxPlot 200-400 nm), 97.77% (220 nm).
-
- 7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
-

-
NaH (2.95 g, 72.4 mmol) was added to an ice-cold solution of 2,3-dihydroxybenzaldehyde (5.0 g, 36 mmol) in anhydrous DMSO (45 mL). Benzyl-3-bromopropyl ether (6.45 mL, 36.2 mmol) was then added and the mixture was stirred at rt for 12 h. The mixture was neutralized using 1 N HCl and then extracted with EtOAc. The organic fraction was washed with H2O and concentrated in vacuo. The residue was purified by flash chromatography (8:2 hexane/EtOAc) to give the title compound as a brown oil: yield 8.40 g (81%).
-
[0891]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.36-7.23 (m, 6H), 7.20-7.16 (m, 2H), 6.98-6.91 (m, 1H), 4.53 (s, 2H), 4.19 (t, J=6.2 Hz, 2H), 3.70 (t, J=6.1 Hz, 2H), 2.19-2.16 (m, 2H).
-

-
[0893]General procedure 6: 3-(3-benzyloxy-propoxy)-2-hydroxy-benzaldehyde (7.6 g, 26 mmol), pyridine (3.42 mL, 42.5 mmol), Tf2O (4.60 mL, 27.9 mmol), and CH2Cl2 (200 mL): yield 8.60 g (77%).
-
[0894]1H NMR (400 MHz, CDCl3) δ (ppm): 10.23 (s, 1H), 7.54-7.47 (m, 1H), 7.43 (t, J=8.0 Hz, 1H), 7.36-7.22 (m, 6H), 4.52 (s, 2H), 4.23 (t, J=6.3 Hz, 2H), 3.71 (t, J=6.1 Hz, 2H), 2.21-2.17 (m, 2H).
-
[0895]General procedure 5: trifluoro-methanesulfonic acid 2-(3-benzyloxy-propoxy)-6-formyl-phenyl ester (8.0 g, 19 mmol), B2pin2 (9.71 g, 38.2 mmol), KOAc (5.71 g, 57.4 mmol), PdCl2(dppf).CH2Cl2 (1.39 g, 1.89 mmol), and anhydrous dioxane (160 mL). Purification: flash chromatography (9:1 hexane/EtOAc): yield 4.80 g (43%)-some pinacol contamination, used without further purification.
-
[0896]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.46 (t, J=7.8 Hz, 1H), 7.41-7.36 (m, 1H), 7.35-7.24 (m, 5H), 7.08 (d, J=7.8 Hz, 1H), 4.50 (s, 2H), 4.10 (t, J=6.3 Hz, 2H), 3.67 (t, J=6.3 Hz, 2H), 2.11 (quin, J=6.2 Hz, 2H), 1.43 (s, 12H).
-

-
General procedure 8: 3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (36 g, 91 mmol), MeNO2 (16.6 g, 273 mmol), NaOH (3.64 g, 83 mmol), H2O (180 mL), and THF (50 mL). Purification: flash chromatography (1:1 hexane/EtOAc). A47 was isolated as a light yellow oil: yield 15.9 g (50%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.05 (s, 1H), 7.44 (t, J=7.8 Hz, 1H), 7.35-7.20 (m, 5H), 7.06 (d, J=7.4 Hz, 1H), 6.88 (d, J=8.2 Hz, 1H), 5.70 (dd, J=9.4, 2.3 Hz, 1H), 5.29 (dd, J=13.7, 2.7 Hz, 1H), 4.53 (dd, J=13.3, 9.4 Hz, 1H), 4.45 (s, 2H), 4.11 (t, J=6.1 Hz, 2H), 3.60 (t, J=6.3 Hz, 2H), 2.04-1.91 (m, 2H); MS (ESI): m/z=356 (M−1, negative); HPLC purity: 99.35% (MaxPlot 200-400 nm), 97.32% (220 nm).
-

-
General procedure 13: A47 (0.50 g, 1.4 mmol), 20% Pd(OH)2/C (0.5 g, 1:1 w/w), AcOH (20 mL), and H2O (0.24 mL). The filtrate was concentrated and treated with 4 N HCl to give the title compound as a colorless solid: yield 0.22 g (47%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.42 (t, J=7.8 Hz, 1H), 6.97-6.90 (m, 1H), 6.86 (d, J=8.2 Hz, 1H), 5.20 (dd, J=9.2, 2.5 Hz, 1H), 4.02 (t, J=6.2 Hz, 2H), 3.54 (t, J=6.2 Hz, 2H), 3.40 (dd, J=13.3, 2.7 Hz, 1H), 2.68 (dd, J=13.1, 9.2 Hz, 1H), 1.88-1.78 (m, 2H); MS (ESI): m/z=238 (M+1, positive).
-

- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A46)

-

-
To a 5° C. solution of compound A (15.0 g, 0.075 mol), B (12.0 ml, 0.075 mol) and triphenylphosphine (19.6 g, 0.075 mol) in 200 ml of anhydrous THF was added DIAD (14.8 ml, 0.075 mol) drop by drop over a period of 15 minutes. The resulting solution was warmed to room temperature over a period of 5 h and the solvent was evaporated in vacuo. The residue was dissolved in 150 ml of EtOAc and the organic layer washed with water, brine and dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) generating 13.0 g (50% yield) of C [3-(3-benzyloxy-propoxy)-2-bromo-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 10.41 (s, 1H), 7.49 (d, J=7.2 Hz, 1H), 7.32-7.25 (m, 6H), 7.08 (d, J=8.0 Hz, 1H), 4.54 (s, 2H), 4.16 (t, J=6.0 Hz, 2H), 3.74 (t, J=5.8 Hz, 2H), 2.19-2.14 (m, 2H).
-

-
Compound C (8.9 g, 0.025 mol), KOAc (7.5 g, 0.076 mol), and bis(pinacolato)diboron (12.9 g, 0.051 mol) were dissolved in 50 ml of dry DMF and degassed for 30 minutes. To this was added PdCl2(dppf).DCM (0.56 g, 0.76 mmol) and the contents were again degassed for 10 minutes and then heated to 90° C. for 4 h. An additional quantity of PdCl2(dppf).DCM (0.2 g, 0.27 mmol) was added and heating was continued for an additional 2 h. The reaction was cooled to RT, filtered through celite and the solvent evaporated in vacuo. The residue was dissolved in DCM, washed with brine and the organic layer dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) provided 5.4 g (53% yield) of D [3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.91 (s, 1H), 7.43 (t, J=7.8 Hz, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.32-7.27 (m, 5H), 7.06 (d, J=8.4 Hz, 1H), 4.49 (s, 2H), 4.08 (t, J=6.0 Hz, 2H), 3.67 (t, J=6.2 Hz, 2H), 2.11-2.08 (m, 2H), 1.44 (s, 12H). ESI+MS m/z, 397 (M+H)+.
-

-
To an ice cold solution of NaOH (0.68 g, 0.017 mol) in 10 ml of water was added a solution of compound D (6.8 g, 0.017 mol) dissolved in 5 ml of THF. After 15 minutes, nitromethane (0.93 ml, 0.017 mol) was added drop by drop and the content stirred at RT overnight. The THF was evaporated under reduced pressure and the contents acidified to pH-3 with 2N HCl. The aqueous layer was extracted with EtOAc several times, and the combined ethyl acetate layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of 10% EtOAc/hexane to 30% EtOAc/hexane) provided 3.7 g (55% yield) of E [7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol] 3.7 g.
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.49 (t, J=7.8 Hz, 1H), 7.34-7.25 (m, 5H), 7.08 (d, J=7.6 Hz, 1H), 6.92 (d, J=8.0 Hz, 1H), 5.71 (d, J=6.4 Hz, 1H), 5.23 (dd, J=13.2, 2.4 Hz, 1H), 4.58-4.53 (m, 1H), 4.47 (s, 2H), 4.12 (t, J=6.2 Hz, 2H), 3.63 (t, J=6.0 Hz, 2H), 2.04-2.00 (m, 2H). ESI-MS m/z, 356 [M−H]−. HPLC purity: 97.12% (MaxPlot 200-400 nm).
- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A46)
-

-
Compound E (6.0 g, 0.016 mol) was dissolved in 50 ml of glacial acetic acid and to it was added Pd(OH)2 on Carbon (20% metal content, 50% weight-wet) (5.2 g) and the content set for hydrogenation in a Parr shaker at 45 psi for 2 h. The reaction was checked for completion and the contents were filtered through Celite. The solvent was evaporated under reduced pressure at ambient temperature to yield a gummy material. To this three times was added 15 ml of dry toluene and evaporated yielding a fluffy solid. Purification was accomplished by preparative HPLC (C18 column, using acetonitrile and 0.1% AcOH/water solution) provided 1.5 g (45% yield) of compound A46 [3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol] with 0.33 mol % acetic acid (by HNMR).
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.52 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.2 Hz, 1H), 6.95 (d, J=8.4 Hz, 1H), 5.29 (dd, J=9.2, 2.4, 1H), 4.12 (t, J=6.2 Hz, 2H), 3.62 (t, J=6.2 Hz, 2H), 3.48 (dd, J=13.2, 2.8 Hz, 1H), 2.80-2.74 (m, 1H), 1.92 (t, J=6.2 Hz, 2H). ESI+MS m/z, 238 [M+H]+. HPLC purity: 95.67% (MaxPlot 200-400 nm) and 96.22% (220 single wavelength).
- (S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A49)
-

-
A solution of 1 (160 g, 962.58 mmol) and triphenylphosphine (277.72 g, 1.1 eq, 1058.83 mmol) was dissolved in dichloromethane (800 mL) and cooled to 0° C. (ice/water). A solution of carbon tetrabromide (351.16 g, 1.1 eq, 1058.83 mmol) in dichloromethane (200 mL) was added dropwise and the mixture was left to stir at rt for 18 h. The dichloromethane solvent was evaporated to obtain a white solid. The solid was treated with an excess of hexanes, stirred for 1 h, filtered off and the solvent was evaporated to yield a crude product. The crude product was purified by silica gel column chromatography using 5-10% ethyl acetate and hexane to obtain 2 (199 g, 91%) as a colorless liquid.
- (3-Benzyloxy)-1-bromo-propane (2)
-

-
To a solution of aldehyde 3 (27.47 g, 1 eq, 198.88 mmol) in 0.5 L of anhydrous DMSO was added sodium tertiary-butoxide (42.3 g, 2.2 eq, 440.31 mmol) portionwise. The reaction mixture was stirred at rt for 30 minutes. A brown color solution was formed. The reaction mixture was cooled to 0° C. and added bromide (56 g, 1.2 eq, 244.41 mmol) dropwise. The mixture was stirred at rt O/N. 90% of aldehyde 3 was converted to product. The reaction mixture was acidified to pH-3 and then extracted into EtOAc and washed with water. The organic layer was concentrated, the product was purified on silica gel column (EtOAc:hexane 80:20), to yield as compound 4 (48 g, 84.31% yield) (viscous oil).
-

-
To an ice cold solution of 4 (48 g, 1.0 eq, 167.72 mmol) in 200 mL of dry DCM was added pyridine (22 mL, 1.62 eq, 272.11 mmol). To the reaction mixture trifluoromethanesulfonic anhydride (33 mL, 1.16 eq, 196.14 mol) was added drop by drop. The mixture was stirred for 3 h at 0° C. The mixture was quenched with 500 mL of 1N HCl. The compound was then extracted into DCM (300 mL) and passed through a small silica gel column and concentrated to give compound 5 (57 g, 82% yield) as a pale yellow thick oil.
-

-
Compound 5 (65 g, 1.0 eq, 155.5 mmol), bis(pinacolato)diboron (86.9 g, 2.2 eq, 342.11 mmol), KOAc (45.7 g, 3.0 eq, 466.5 mmol) were mixed together and 600 mL of dioxane was added. The mixture was degassed with N2 for 30 minutes and PdCl2(dppf).DCM (5.7 g, 0.05 eq, 7.77 mmol) was added. The resulting slurry was heated to 90° C. overnight. The solvent was evaporated, EtOAc was added and then filtered through a pad of Celite. The organic layer was then washed with water (2×150 mL) and the solvent was evaporated. Column chromatography using 15% EtOAc/hexanes gave compound 6 (37.1 g, 61% yield).
-

-
A solution of compound 6 (36 g, 1.0 eq, 90.91 mmol) in 50 mL of THF was cooled to 0° C. Nitromethane (16.6 g, 3.0 eq, 272.72 mmol) was added, followed by an aqueous solution of NaOH (3.64 g in 180 mL of H2O). The reaction mixture was stirred at room temperature overnight. The starting material disappeared. The cyclization was afforded by adding 1N HCl until the solution was acidified and then extracted into EtOAc. The EtOAc was evaporated and the mixture was triturated with water and decanted. Column chromatography using 50% EtOAc/hexanes gave compound A47 (15.9 g, 50% yield).
-

-
4.82 g of (A47) was resolved via chiral HPLC using CHIRALPAK ADH column and CO2:methanol (86:14) as eluent (25° C. UV detection was monitored at 230 nm. Two peaks, (S)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol and (R)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol were collected and evaporated to yellow oils. Analysis of the pooled fractions using a CHIRALPAK ADH 4.6 mm ID×250 mm analytical column and the same mobile phase provided the (S) enantiomer [0.7 g (29% yield)] with a retention time of 6.11 min and a 98.2% ee. The (R) enantiomer [1.0 g (41% yield)] had a retention time of 8.86 min and a 99.6% ee.
-

-
(A47) (550 mg, 1.57 mmol) was dissolved in 15 mL of glacial acetic acid. 280 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 128 mg of the acetate salt of (A49). The acetate salt was treated with 10 mL of 2N HCl and stirred for 3 hours. The material was lyophilized overnight to obtain 93 mg of the hydrochloride salt of (A49) (Yield 22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.82 (dd, J=13.3, 9.0 Hz, 1H), 1.95-1.83 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 98.74% (MaxPlot 200-400 nm), 98.38% (220 nm); Chiral HPLC=95.14% ee.
- (R)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A50)
-

-
(R)-7-(3-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (0.70 g, 2.0 mmol) was dissolved in 20 mL of glacial acetic acid. 350 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 65 mg of pure compound. After purification, this acetate salt was combined with material from another reaction. This product was treated with 2N HCl (10 mL) and stirred for 3 h at rt. The material was lyophilized overnight to obtain 74 mg of the hydrochloride salt of (A50) (Yield 14%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.83 (dd, J=13.3, 8.6 Hz, 1H), 1.94-1.82 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 99.12% (MaxPlot 200-400 nm), 98.74% (220 nm); Chiral HPLC=98.82% ee.
https://pubs.acs.org/cen/coverstory/89/8912cover3.html
https://www.yumpu.com/en/document/view/34463506/the-discovery-of-gsk2251052-a-first-in-class-boron-anacor
| US20040203094 * | Sep 20, 2002 | Oct 14, 2004 | Martinis Susan A. | Eucyl-tRNA synthetases and derivatives thereof that activate and aminoacylate non-leucine amino acids to tRNA adaptor molecules |
| US20070155699 * | Aug 16, 2006 | Jul 5, 2007 | Anacor Pharmaceuticals | Boron-containing small molecules |
| US20090227541 * | Jun 19, 2008 | Sep 10, 2009 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
Anacor
BIG TEAM Hernandez, front row, fifth from left, poses during a research meeting at Naeja’s headquarters.

Dr. R. G. Micetich’s research career began in 1963 as a Research Scientist with R & L Molecular Research Ltd. (established by Dr. R. U. Lemieux). This company later became Raylo Chemicals Ltd. Dr. Micetich served as the Research Director (Pharmaceutical Research) of Raylo. During the period from 1963 to 1980 Dr. Micetich’s group was involved in pharmaceutical research and process development work in antibiotics and in NSAI’s (non-steroidal anti-inflammatory agents). This work produced a drug “Mofezolac” – a NSAI which is now marketed in Japan by the Japanese company “Yoshitomi”. Market ~ U.S. $60 million.
In 1980, Dr. R. G. Micetich joined the Faculty of Pharmacy, University of Alberta as an Adjunct Professor working on projects for international big Pharma companies. The work with Taiho Pharmaceutical Company in Japan has produced another drug – a beta-lactamase inhibitor – “TAZOBACTAM” which is now marketed worldwide. This drug now produces annual sales of over US$ 1 billion.
In 1987, Dr. Micetich established a joint venture research company with Taiho, Japan called SynPhar. SynPhar had numerous patents worldwide in various therapeutic areas and many compounds and classes of compounds at various stages of development up to late preclinical.
In view of the significant growth opportunities for SynPhar and in response to the changing international market place for pharmaceuticals, Dr. Micetich acquired and transferred all the assets including intellectual property, equipment and fixtures from SynPhar to NAEJA Pharmaceutical Inc. in 1999. NAEJA is a private Albertan company, founded by the Micetich family which from an initial staff in August 1999 of 40, has grown to 130 and is still growing. NAEJA is a completely self-supporting private company with no venture capital, nor private, nor government funding. The majority of NAEJA employees hold Ph.D.’s. NAEJA has collaborative agreements with pharmaceutical companies around the world. Based on its own intellectual property, NAEJA also has a number of co-development agreements with biotech companies worldwide. Dr. Micetich laid the seeds of foundation for NAEJA and the company continues after his passing, building his legacy.
Dr. R. G. Micetich boasted over 100 publications in well know scientific journals and composed over 100 patents taken out in many countries…………..http://www.bioalberta.com/ron-micetich
more……….
RONALD G. MICETICH (1931-2006): A Scientific Career Ronald Micetich was born in Podanur, Coimbatore (South India). Following receipt of B.Sc. Honors (Chemistry, Loyola College, Madras) and M.A. (Chemistry, Madras University) degrees in India, Ron obtained a Ph.D. (Organic Chemistry, University of Saskatchewan, Canada) in 1962. Ron initiated his interest in microbiology while he was a postdoctoral fellow at the National Research Council of Canada. During the period 1963-1980, Ron held a number of industrial appointments where he rapidly advanced his industrial scientific career (research scientist → associate research director → acting research director → director pharmaceutical / agricultural research) at Raylo Chemicals in Edmonton, Alberta. In 1981 Ron joined the Faculty of Pharmacy & Pharmaceutical Sciences at the University of Alberta as an Adjunct Professor at which time a highly successful drug development program was established with Taiho Pharmaceuticals. This joint industrial collaboration led to the birth of SynPhar with Dr. Micetich as Chairman of the Board, President, CEO and Research Director (1987-1999). Ron, again as Chairman of the Board, CEO and Research Director, established NAEJA (North America, Europe, Japan, Asia) Pharmaceuticals in 1999 with a rollover of assets, including staff, equipment and intellectual property, from SynPhar Laboratories. What began as a full fledged pharmaceutical company with an extensive intellectual property portfolio and a proven track record evolved into an internationally respected pharmaceutical outsource service provider. NAEJA has carved a unique niche in the outsource industry offering extensive discovery experience and expertise. Today, NAEJA has over 120 staff that consists of over 90% scientists holding PhD degrees.,………….see link below
[PDF]RONALD G. MICETICH – University of Alberta – Journal …
by T Yajima – 2008

Dr.Muhammed Majeed with Dr. Ronald Micetich, CEO, Naeja Pharmaceuticals, Edmonton Canada’.


Christopher Micetich
Founder, President & CEO, Board Chairman
Fedora Pharmaceuticals Inc.
See us at: http://www.fedorapharma.com
Fedora Pharmaceuticals has developed a family of beta-lactamase
inhibitors designed to have activity against pathogens containing all
four classes of beta-lactamases. These promising novel beta-lactamase
inhibitors, used in combination with various beta-lactam antibiotics to
treat those antibiotic infections currently resistant to therapy, have
recently been licensed to Swiss-based pharmaceutical giant, Hoffman La
Roche Ltd. in what is being touted as one of the largest biotech
licensing deals ever signed in Canadian history!Fedora Pharmaceuticals in Canada and Meiji Seika in Japan, with shared world-wide rights, have teamed and jointly entered into this significant tripartite agreement with Roche. Under the terms of the agreement, Roche will obtain exclusive rights from both companies to develop and commercialize the agent worldwide excluding Japan. Fedora and Meiji will receive from Roche; an upfront payment, development, regulatory and sales event milestone payments in addition to royalties on sales of products. While the details of the amounts have not been disclosed a total deal value of US$750 Million in addition to royalties has been announced.
Fedora was founded in 2012 and is headquartered in Edmonton, Alberta, Canada.

President & CEO, Founder
Naeja
NAEJA
Pharmaceuticals Inc. is a privately controlled pharmaceutical CRO
specializing in early stage drug discovery research with particular
expertise in the area of Medicinal Chemistry. NAEJA employs highly
skilled PhD scientists recruited from around the globe.
The company boasts a very long and successful track record of rapidly
advancing drugs through discovery into the clinic. Several drugs in
latter stages of clinical development and on the market today have
originated from NAEJA. Most recently is Fedora Pharmaceuticals US $750M
licensing deal to Hoffman La Roche Ltd that originated from the
laboratories of NAEJA.As a privately controlled company, NAEJA are very responsive to client’s needs offering many years of drug discovery experience to successfully find ways to advance research programs in a timely, cost effective and efficient manner. NAEJA’s longstanding track record is a testimony in itself!
B1(c2c(cccc2OCCCO)[C@H](O1)CN)O
4 Crisaborole, AN 2728
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis......Anacor Pharmaceuticals, Inc.Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.
Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
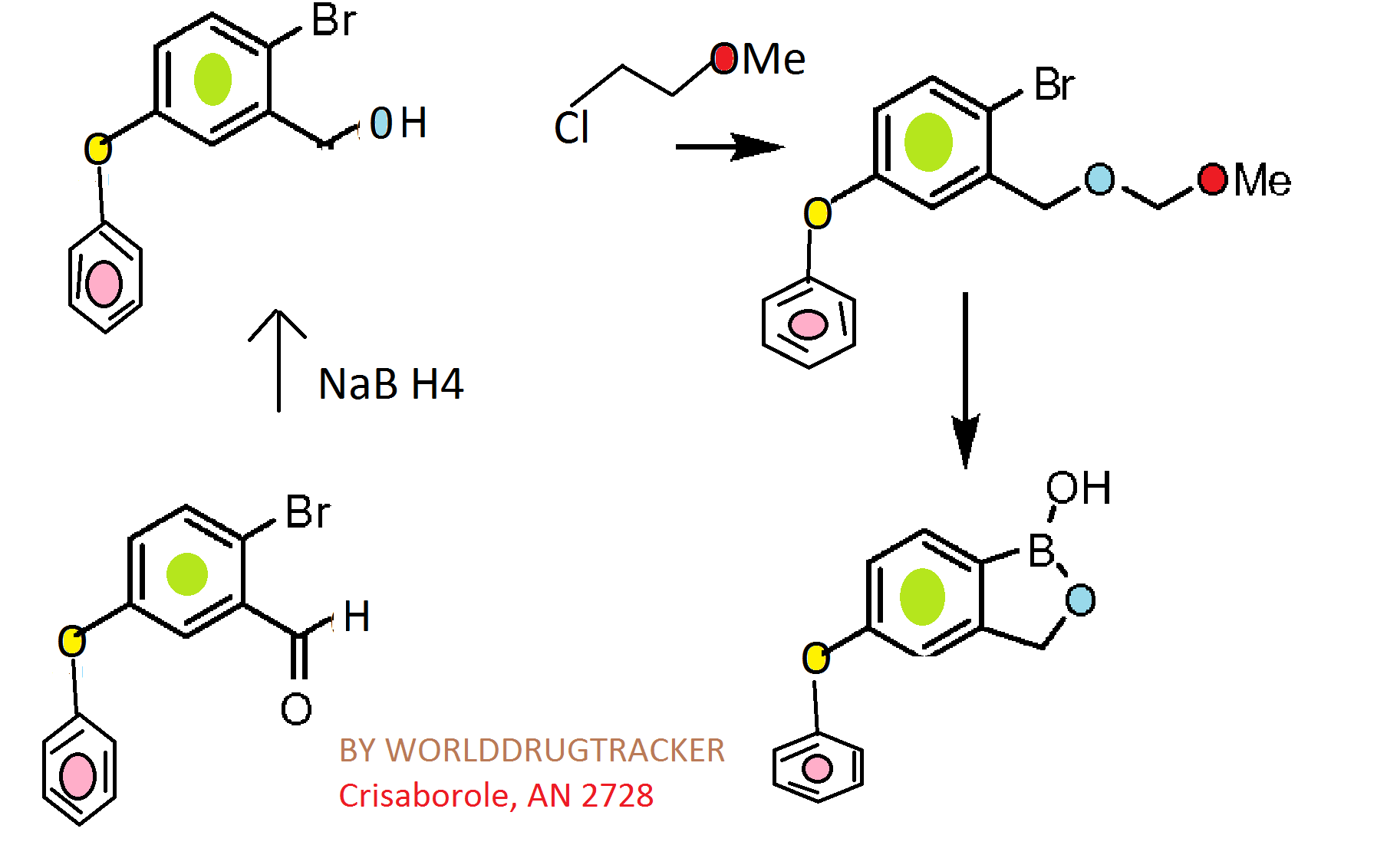
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi,
THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M
NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)

- (a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
- (b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer's disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women's Hospital, Inc. | Use of gelsolin to treat infections |
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
... Francisco, CA Mar. 6-10, 2009. 6, "AN2728 ... Francisco, CA Mar. 6-10, 2009. 7 , "AN2728 ... Kyoto, Japan, May 14-18, 2008. 10, "AN2728 ...
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- .... UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
... Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, "AN2728 ... 7, "AN2728 ... Francisco, CA May 6-10, 2009. 10, "AN2728 ...
|
... from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, ...
|
"AN2728" is the compound 4-(l-hydroxy-l,3-dihydro-2 ... GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, ...
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- .... UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
5 AN-2718
AN-2718,
2,1-Benzoxaborole, 5-chloro-1,3-dihydro-1-hydroxy-
5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole5-Chloro-2,1-benzoxaborol-1(3H)-ol
CAS 174672-06-1
UNII: 810U6C2DGG
MW 168.3864, MF C7 H6 B Cl O2
MP...150-154 °C [WO 9533754]MP 142-144 °C [ jmc 2006, 49(15) 447]
M.p. 147- 149 °C. WO2013050591
Anacor Pharmaceuticals Inc, INNOVATOR
Onychomycosis is a disease of the nail caused by yeast, dermatophytes, or other molds, and represents approximately 50% of all nail disorders. Toenail infection accounts for approximately 80% of onychomycosis incidence, while fingernails are affected in about 20% of the cases. Dermatophytes are the most frequent cause of nail plate invasion, particularly in toenail onychomycosis. Onychomycosis caused by a dermatophyte is termed Tinea unguium. Trichophyton rubrum is by far the most frequently isolated dermatophyte, followed by T. mentagrophytes. Distal subungual onychomycosis is the most common presentation of tinea unguium, with the main site of entry through the hyponychium (the thickened epidermis underneath the free distal end of a nail) progressing in time to involve the nail bed and the nail plate. Discoloration, onycholysis, and accumulation of subungual debris and nail plate dystrophy characterize the disease. The disease adversely affects the quality of life of its victims, with subject complaints ranging from unsightly nails and discomfort with footwear, to more serious complications including secondary bacterial infections.
Many methods are known for the treatment of fungal infections, including the oral and topical use of antibiotics (e.g., nystatin and amphotericin B), imidazole anti-fungal agents such as miconazole, clotrimazole, fluconazole, econazole and sulconazole, and non-imidazole fungal agents such as the allylamine derivatives terbinafme and naftifϊne, and the benzylamine butenafine. However, onychomycosis has proven to be resistant to most treatments. Nail fungal infections reside in an area difficult to access by conventional topical treatment and anti-fungal drugs cannot readily penetrate the nail plate to reach the infection sites under the nail. Therefore, onychomycosis has traditionally been treated by oral administration of anti-fungal drugs; however, clearly this is undesirable due to the potential for side effects of such drugs, in particular those caused by the more potent anti-fungal drugs such as itraconazole and ketoconazole. An alternative method of treatment of onychomycosis is by removal of the nail before treating with a topically active anti-fungal agent; such a method of treatment is equally undesirable. Systemic antimycotic agents require prolonged use and have the potential for significant side effects. Topical agents have usually been of little benefit, primarily because of poor penetration of the anti-fungal agents into and through the nail mass.
AN-2718 is a topical benzoxaborole compound that has a high level of nail penetrance [51]. Initial data have suggested that it may be more effective than tavaborole for T. rubrum and T. mentagrophytes [55]. It has recently completed Phase I clinical trials (NCT00781664) [56].
picked from
- 51. Hui X, Baker SJ, Wester RC, In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci 2007;96(10):2622-31 [CrossRef], [PubMed], [Web of Science ®]
- 55. Mao W, Seiradake E, Crepin T, AN2718 has Broad Spectrum Antifungal Activity Necessary for the Topical Treatment of Skin and Nail Fungal Infections. J Am Acad Dermatol 2007:56(2 Suppl):AB124
- 56. ClinicalTrials.gov. Cumulative Irritation Test. Available from: http://clinicaltrials.gov/ct2/show/NCT00781664 [Cited 11 December 2011]

Reduction of 2-bromo-5-chlorobenzoic acid with BH3/THF in THF gives 2-bromo-5-chlorobenzyl alcohol , which is protected as the methoxymethyl derivative by treatment with MOM-Cl in the presence of DIEA in CH2Cl2. Metalation and boronylation of aryl bromide either with t-BuLi or BuLi (1,2,3,4) and (i-PrO)3B (4) or B(OMe)3 in THF affords the title compound .
Alternatively, reaction of 3-chlorobenzaldehyde with p-toluenesulfonylhydrazide in MeOH provides tosyl hydrazone which undergoes thermal decomposition in the presence of BBr3 and catalytic FeCl3 in refluxing CH2Cl2, followed by heating with aqueous NaOH to produce the title oxaborole compound .
You can construct from this................
 OH A
OH A(2-bromo-5-chloro-phenyl)methanol (A)
 B
Bl-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
 1
15-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
WO 9533754https://www.google.com/patents/WO1995033754A1?cl=en
Example 1 Preparation of 5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole (Method B).
a)
Preparation of 3-chlorobenzaldehyde tosyl hydrazide
A solution of 3-chlorobenzaldehyde (15.56 parts; 0.109M;
Aldrich) in methylated spirits (40 ml) was added slowly at below 10°C to a stirred suspension of p-toluene-sulphonylhydrazide (20.7 parts;
0.108M) in methylated spirits (150 ml). The reaction mass was then stirred at 20 to 25°C for 1 hour and then heated at 60-70°C for 1M hours when the reactants and products dissolved. The solvent was then removed by rotary evaporation and the product was obtained as a solid which was slurried with ether and washed with n-hexane. Yield = 27.2 parts (81.5% theory) mpt 122-3°C. Elemental analysis
Theory 54.5%C; 4.2%H; 9.1%N
Found 54.5%C; 4.3%H; 9.1%N
Proton NMR (CDCl3:ppm)
8.5, s, 1H(-NH-); 7.9, d, 2H(Tosyl aromatic); 7.7,s, 1H(CH=N); 7.5, s, 1H (aromatic); 7.2-7.4, m, 5H(Tosyl aromatic); 2.3, s, 3H(-CH3) b) Preparation of title compound
A suspension of anhydrous ferric chloride catalyst (0.75 parts, Fisons) in dry dichloromethane (20 ml) was added at 20 to 25°C simultaneously with boron tribromide (25 parts, 0.1M, Aldrich) in dry dichloromethane (100 mis) to a stirred suspension of the hydrazide from a) above (10.18 part, 0.033M) in dry dichloromethane (160 mis) under a nitrogen blanket. The reactants were then stirred under reflux and the evolved hydrogen bromide trapped under aqueous sodium hydroxide. After 3 hours stirring at reflux, the reactants were allowed to stand at 20- 25°C for 48 hours and then stirred under reflux for a further 4 hours. The reaction mass was then cooled and the solvent removed by rotary evaporation. The solid obtained was then stirred under reflux with 2N sodium hydroxide solution (160 ml) for 3 hours. The brown aqueous suspension was extracted with dichloromethane (50 ml), screened and then acidified to about pH 2 by addition of 2N hydrochloric acid. The solid was filtered, slurried with dichloromethane (400 ml) and then washed with a saturated solution of sodium bicarbonate followed by water.
Yield = 24 parts (43% theory). The solid was slurried in hot
dichloromethane and filtered to give 0.36 parts oxaborole mp 140-45°C. The dichloromethane solution was cooled and the solid filtered giving a further 0.35 parts oxaborole mp 146-8°C. The solids were combined and recrystallised from methylated spirits.
Yield = 0.51 parts (9.2% theory) mp 150-4°C.
Elemental Analysis
Theory 49.8%C, 3.5%H, 21.06%C1
Found 49.5%C, 3.5%H, 21.0%C1
Proton NMR (CDCl3) ppm
9.3, s, 1H(-OH); 7.5, d, s, d, 3H(aromatic);
5.0, s, 2H(-CH2-O).
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en

Analytical data for exemplary compounds of structure I are provided below.
4.2. a 5-Chloro-1.3-dihydro-l -hvdroxy-2J-benzoxaborole (Cl) M.ρ. 142-15O0C. MS (ESI): m/z = 169 (M+l, positive) and 167 (M-I, negative). HPLC (220 nm): 99% purity. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.71 (d, J = 7.8 Hz, IH), 7.49 (s, IH), 7.38 (d, J = 7.8 Hz, IH) and 4.96 (s, 2H) ppm.
PAPER
J. Med. Chem., 2006, 49 (15), pp 4447–4450
DOI: 10.1021/jm0603724
http://pubs.acs.org/doi/abs/10.1021/jm0603724?source=chemportCOMPD IS 19d
5-Chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (19d) This compound was made from 18d in the same manner as compound 19b (triturated with hexane, 75% yield):: mp 142-144°C. 1 H NMR (300MHz, DMSO-d6) δ (ppm) 4.96 (s, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 9.30 (s, 1H); ESI-MS m/z 167 (M-H)- ; HPLC purity 99.0%; Anal (C7H6BClO2 ⋅ 0.1H2O) C, H
precursor 18d
2-Bromo-5-chloro-1-(methoxymethoxymethyl)benzene (18d) To a solution of 2-bromo-5-chlorobenzoic acid (5.49 g, 23.3 mmol) in anhydrous THF (70 mL) under nitrogen was added dropwise a BH3 THF solution (1.0 M, 55 mL) at 0o C and the reaction mixture was stirred overnight at room temperature. Then the mixture was cooled on an ice bath and MeOH (20 mL) was added dropwise to decompose excess BH3. The resulting mixture was stirred until no bubble was released and then 10% NaOH (10 mL) was added. The mixture was concentrated and the residue was S6 mixed with water (200 mL) and extracted with EtOAc. The residue from rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 2-bromo-5-chlorobenzyl alcohol as a white solid (4.58 g, 88%): 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57 (d, J = 8.7 Hz, 1H), 7.50-7.49 (m, 1H), 7.28-7.24 (m, 1H), 5.59 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H). 2-Bromo-5-chlorobenzyl alcohol obtained above was dissolved in CH2Cl2 (150 mL) and cooled to 0o C on an ice bath. To this solution under nitrogen were added in sequence i-Pr2NEt (5.4 mL, 31 mmol) and chloromethyl methyl ether (2.0 mL, 26 mmol). The reaction mixture was stirred overnight at room temperature and washed with NaHCO3-saturated water and then brine. The residue after rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 18d (4.67 g, 85%) as a colorless oil: 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 3.30 (s, 3H), 4.53 (s, 2H), 4.71 (s, 2H), 7.32 (dd, J = 8.4, 2.4 Hz, 1H), 7.50 (dd, J = 2.4, 0.6 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H).
PATENT
http://www.google.com/patents/WO2013050591A2?cl=en
EXAMPLES Example 1 : Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
Step 1 : Preparation of (2-bromo-5-chloro-phenyl)methanol (A)
Step 2: Preparation of l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
Step 3: Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
http://www.google.fr/patents/WO2010110400A1?cl=en&hl=fr
Reference Example 187
5-chloro-2,1-benzoxazine ball roll -1 (3H) - All

PATENT
http://www.google.com/patents/WO2008070257A2?cl=en
5-Fluoro-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole (Cl)
1-Hydroxy-dihydrobenzoxaboroles, such as Cl, were synthesized as shown in Scheme 1. The protected o-bromobenzyl alcohol derivative (18), prepared from 16 or 17, was converted into the corresponding phenyl boronic acid. Deprotection of the methoxymethyl ether using hydrochloric acid followed by spontaneous cyclization gave the target compounds.
Scheme 7

References
- Hui, Xiaoying; Journal of Pharmaceutical Sciences 2007, 96(10), Pg2622-2631
- Baker, Stephen J.; Journal of Medicinal Chemistry 2006, 49(15), Pg4447-4450
- Austin, Peter William; WO 9533754 A1 1995 CAPLUS
| US5880188 * | 26 May 1995 | 9 Mar 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US6083903 * | 16 May 1995 | 4 Jul 2000 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| WO2005013892A2 | 15 Jun 2004 | 17 Feb 2005 | Tsutomu Akama | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| Reference | ||
|---|---|---|
| 1 | * | Austin et al., 1996, CAS: 124:234024. |
| 2 | * | fungicide: definition from Answre.com, 1998. |
| 3 | S. J. Baker, et al., "Progress on New Therapeutics for Fungal Nail Infections,"Annual Reports in Medicinal Chemistry, 40:323-335 (2005). | |
| 4 | Sudaxshina Murdan, "Drug Delivery to the Nail Following Topical Application," International Journal of Pharmaceutics, 236:1-26 (2002). | |
B1(c2ccc(cc2CO1)Cl)O
6 AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref.......... Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
7 coming................
8 coming..............
Plecanatide, also called Trulance, a synthetic analogue of uroguanylin, is a guanylate cyclase-C (GCC) agonist being developed by Synergy Pharmaceuticals for the treatment of gastrointestinal disorders, such as chronic idiopathic constipation (CIC) and irritable bowel syndrome with constipation (IBS-C). In January 2017, an oral formulation of plecanatide received its first global approval in the USA for the treatment of adult patients with CIC. Plecanatide
ReplyDeleteHERPES INFECTION CURE IS 100% AVAILABLE WITH THE HELP OF DR OGBERAESE HERBAL MEDICINE.Joy and happiness is all i can see around ever since i came in contact with this great man Called Dr Ogberaese. i complained bitterly to him about me having herpes only for him to tell me it’s a minor stuff. He told me he has cured thousands of people but i did not believe until he sent me the herbal medicine and i took it as instructed by this great man, only to go to the hospital after two weeks for another test and i was confirmed negative. For the first time in four years i was getting that result. i want to use this medium to thank this great man. His name is Dr Ogberaese , i came in with his email through a friend in USA called Edith Rose and ever since then my live has been full with laughter and great peace of mind. i urge you all with herpes or HSV1&2 to contact him if you are willing to give him a chance. you can contact him through his email drogberaese54@gmail.com or you can also WhatsApp him +2348073818653
ReplyDelete